Design of Biased Agonists Targeting the Apelin Receptor
Cardiovascular diseases remain the leading cause of mortality in the Western world, with heart failure experiencing the most rapid increase over the past decade. Taking statistics from the United States CDC as an example, currently, about 6.2 million people in the U.S. have been diagnosed with heart failure, and in 2018, there were 379,800 death certificates (accounting for 13.4%) that mentioned heart failure.
Heart failure is defined as the heart's inadequate capacity to pump blood effectively due to systemic demands, leading to symptoms of premature fatigue, difficulty in breathing, and edema. It is commonly caused by a series of prevalent conditions, including long-standing hypertension, acute myocardial infarction or ischemia related to coronary artery disease, valve dysfunction and stenosis, infective myocarditis, congenital malformations, familial hypertrophic and dilated cardiomyopathy, as well as diabetic cardiomyopathy, among others. Most of these causes initially provoke a stage of cardiac hypertrophy, during which individual cardiomyocytes respond by increasing in length and width to enhance the heart's pumping function and reduce ventricular wall tension (resulting in a "compensatory hypertrophy" state). However, over the long term, cardiac hypertrophy predisposes individuals to the development of heart failure, arrhythmias, and sudden death.
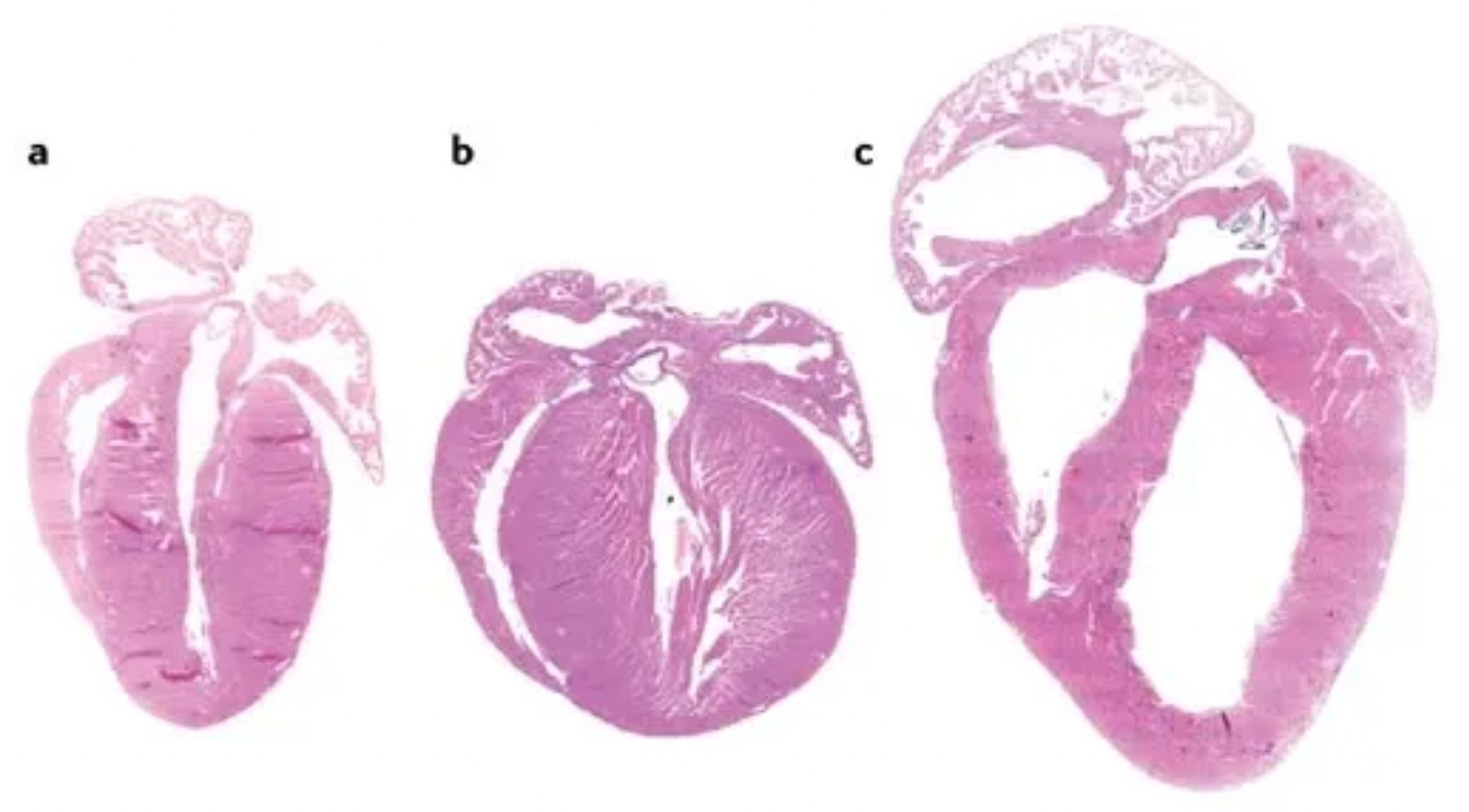
The figure depicts different phenotypes of cardiac hypertrophy, where (a) represents the heart of an adult wild-type mouse; (b) shows a transgenic mouse heart exhibiting a phenotype characterized by concentric hypertrophy, which refers to the uniform thickening of the myocardial layers while the size of the ventricular chamber remains relatively unchanged; and (c) displays a transgenic mouse heart presenting with eccentric hypertrophy and ventricular dilation, wherein the eccentric hypertrophy involves non-uniform thickening of the myocardium, often accompanied by an unequal ventricular wall thickness and enlargement of the ventricular chamber.
The most direct initiating stimuli for cardiac hypertrophy can be categorized broadly into two main classes: biomechanical and stretch-sensitive mechanisms, and neurohumoral mechanisms associated with the release of hormones, cytokines, chemokines, and peptide growth factors. Ligands are perceived by cardiomyocytes through an array of membrane-bound G protein-coupled receptors (GPCRs), receptors with intracellular tyrosine kinase domains, receptors possessing intracellular serine/threonine kinase domains, and gp130-associated receptors. These signaling pathways coordinate hypertrophic growth directly by altering gene expression in the nucleus and by increasing the rate of protein translation in the cytoplasm as well as decreasing the rate of protein degradation.
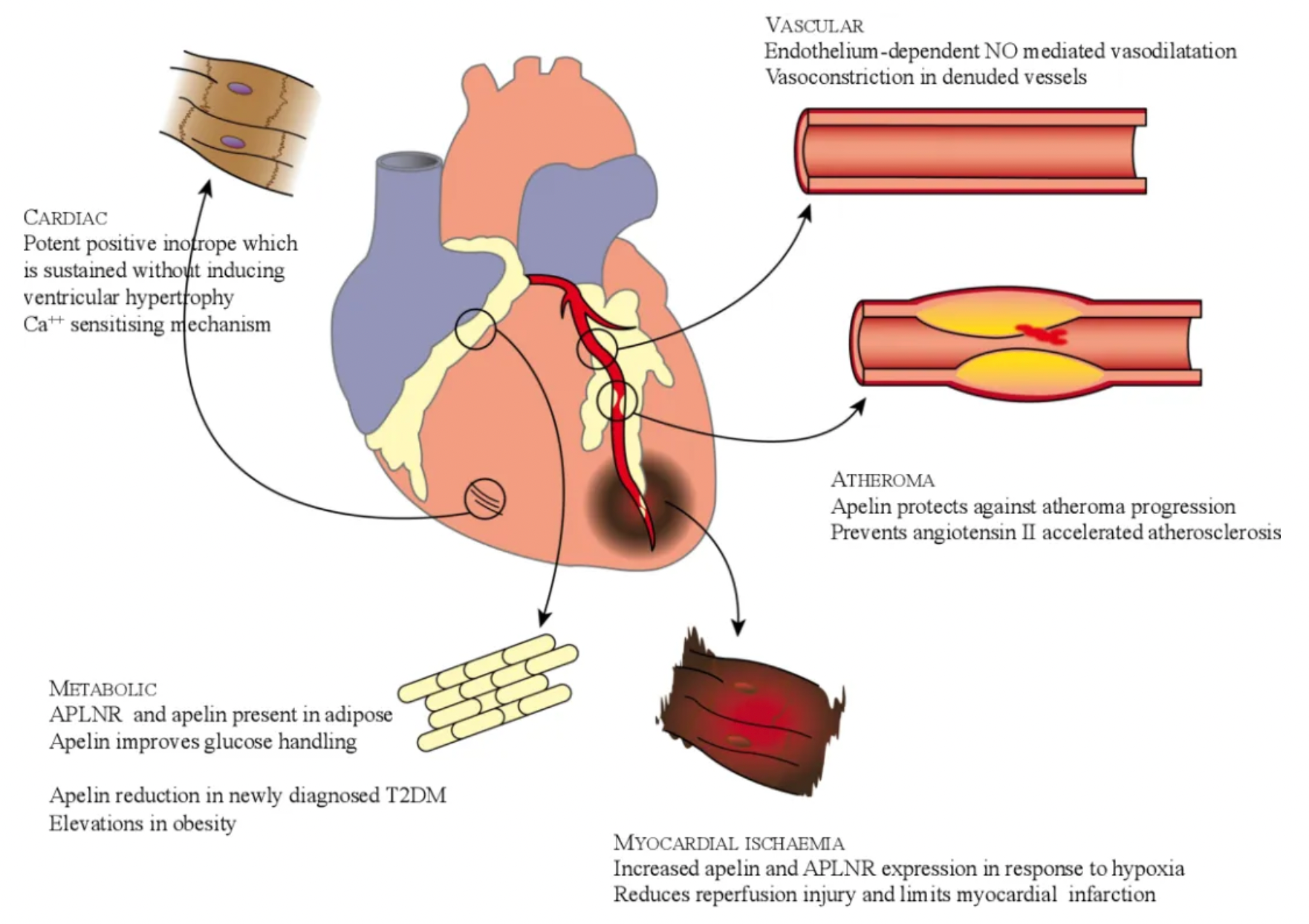
The Apelin receptor (APLNR) plays a crucial role in the cardiovascular system, involved in physiological effects that include promotion of myocardial contractility, enhancement of left ventricular ejection, vasodilation, promotion of diuresis, and reduction of systemic blood pressure. Apelin is the endogenous ligand of APLNR and as a balanced agonist, activates both the G protein pathway and the β-arrestin pathway of APLNR. Apelin-activated APLNR signals through Gαi, having a positive effect on myocardial contractility, and exhibits vasodilatory activity opposing arterial atherosclerosis induced by angiotensin II. Administration of apelin can inhibit the progression of cardiac hypertrophy while apelin knockout mice show increased susceptibility to heart failure. However, the β-arrestin pathway of APLNR is associated with adverse cardiac hypertrophic effects.
On March 14, Professors Zhang Yan from Zhejiang University and Zhang Yan from Peking University jointly published a research paper in the journal "Cell," titled "Structure-based design of non-hypertrophic apelin receptor modulator." The study reveals a structure-guided strategy for biased drug optimization and has led to the development of two biased agonist drug candidates targeting the APLNR receptor.

Researchers decoded the high-resolution structures of the human APLNR bound to agonists with different pharmacological characteristics, uncovering the critical determinants for signal bias of APLNR. Based on this understanding, they rational designed two biased agonists, WN561 and WN353, that selectively activate the G protein pathway. Experiments demonstrated that WN561 has a significant cardioprotective effect, effectively combating cardiac hypertrophy with reduced side effects. The study detailed the experimental methods, sources of reagents, and data storage information, providing a valuable resource for subsequent research.
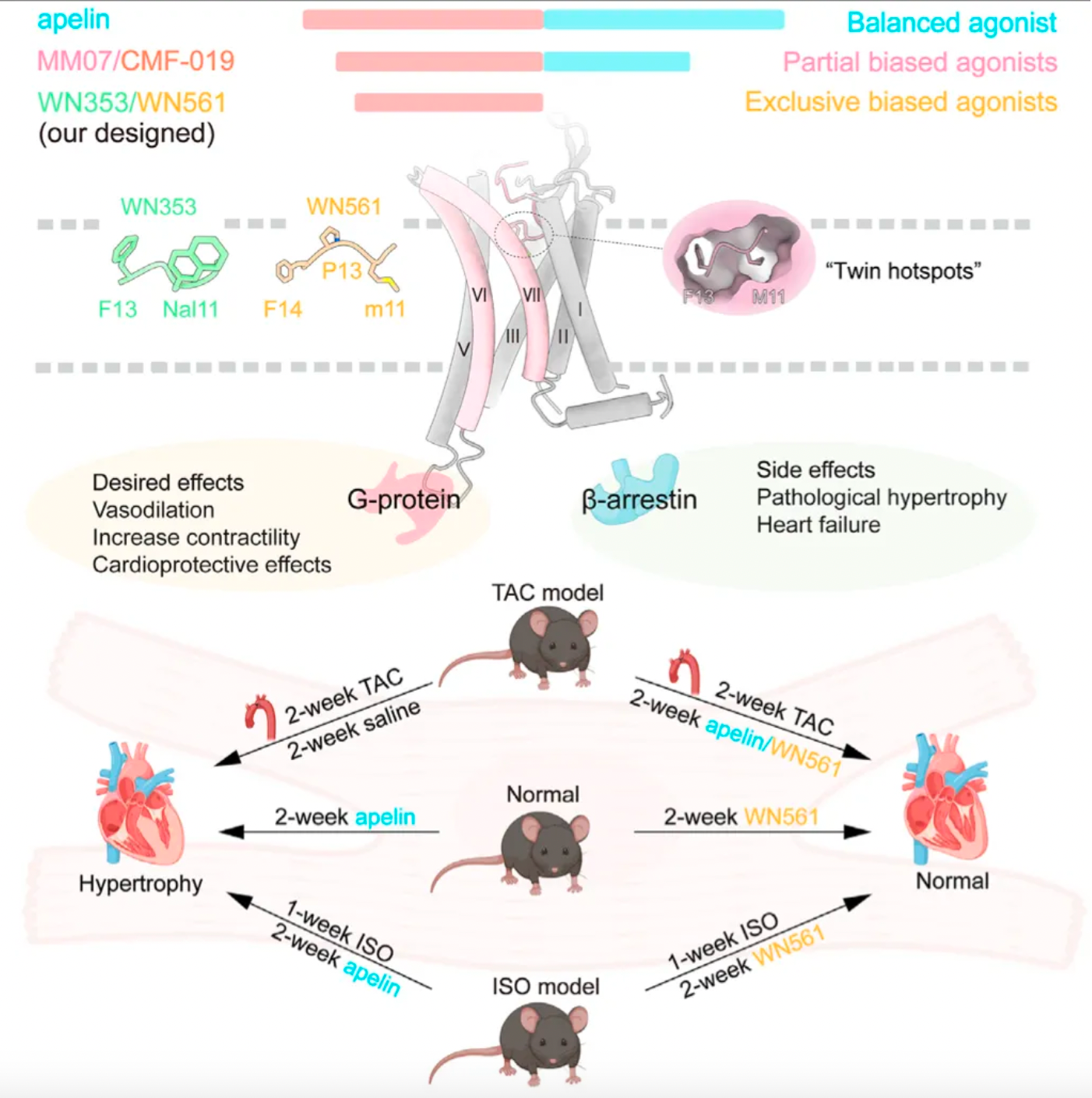
Researchers first compared the pharmacological characteristics of three different APLNR agonists, with particular focus on the activation features of these molecules in the downstream G protein signaling pathway and the β-arrestin signaling pathway. The results showed that CMF-019's efficacy in activating G proteins was comparable to that of apelin, but significantly reduced the recruitment of β-arrestin. MM07, although about 100 times less potent in activating G proteins, led to a roughly 1000-fold decrease in β-arrestin recruitment. These findings indicate that both MM07 and CMF-019 exhibit a strong bias towards the G protein pathway, which is consistent with the results of previous pharmacological studies.
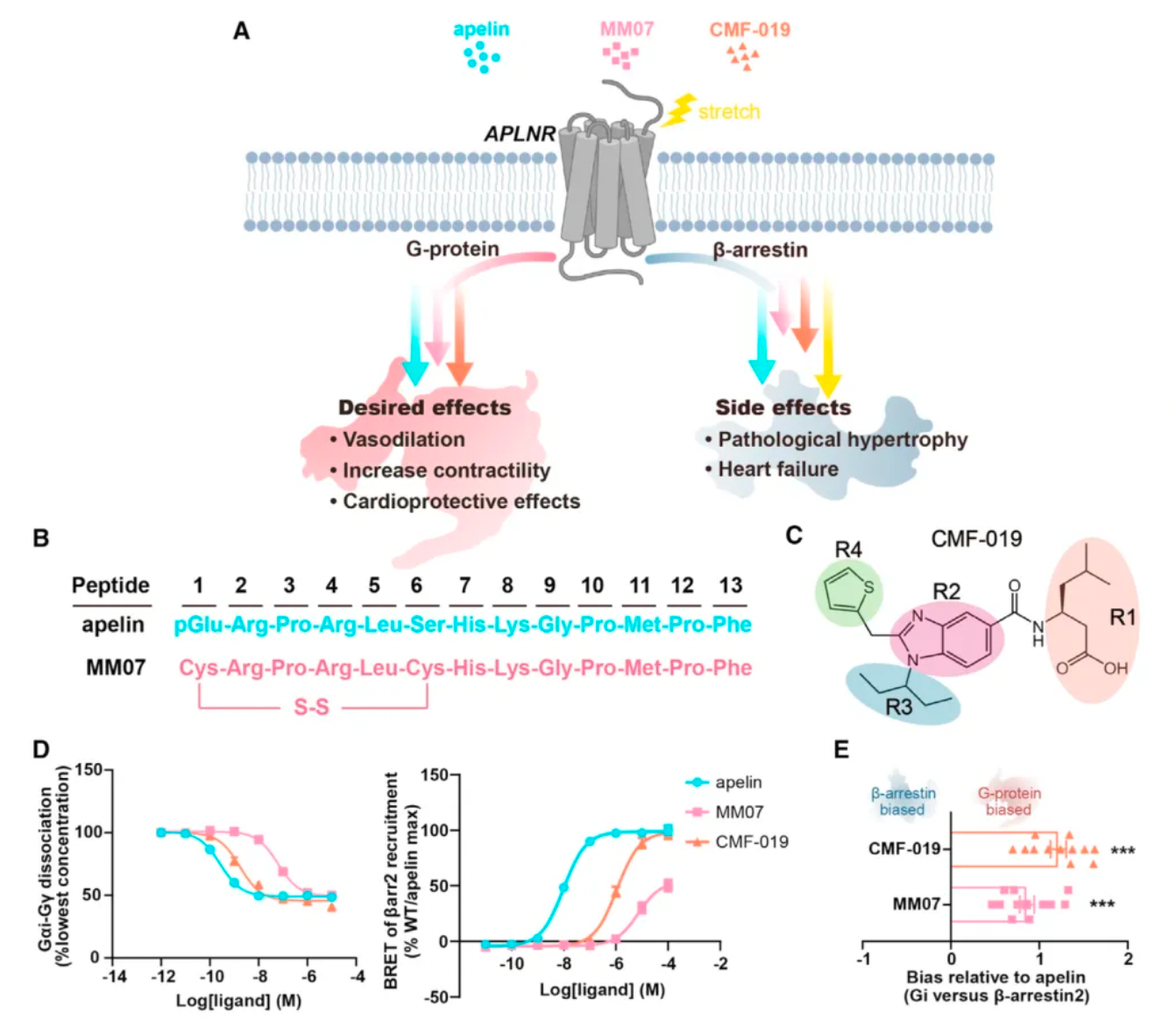
Subsequently, researchers used cryo-electron microscopy to study the complex structures of the three agonists with APLNR, and compared the downstream signaling pathways of the three agonists with multiple receptor mutants. The results revealed substantial differences in the signaling coupling mechanisms between the three molecules and the different receptor mutants due to the variations in the binding modes of the three ligand-receptor complexes.
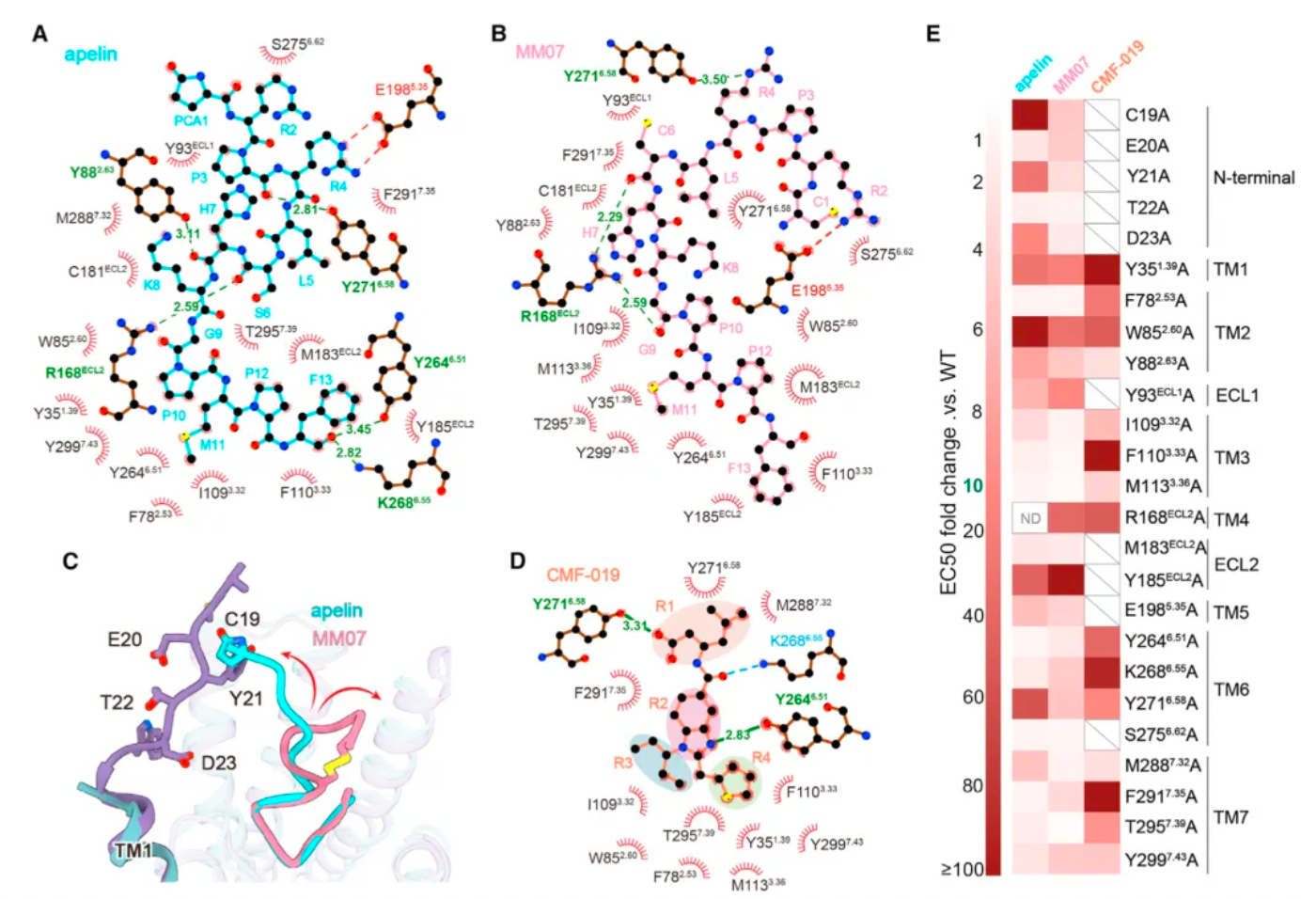
Even more intriguing is that after researchers identified mutations in a portion of the receptor's amino acids, the ligand exhibited a preference for the β-arrestin pathway in its agonist activity across three downstream signaling pathways of the receptor. Based on this, the study identified two potential hotspots through mutagenesis experiments that are very critical for the ligand's G protein-biased agonism.
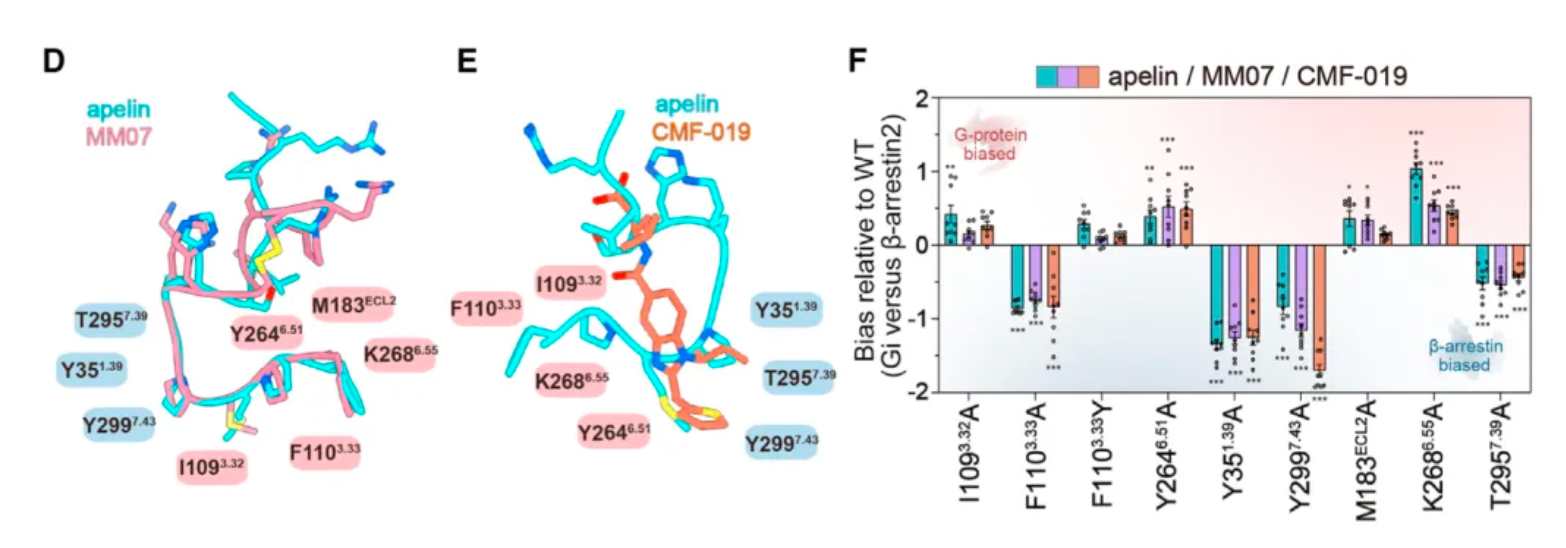
Researchers believe that by structurally modifying ligands targeting these hotspots, there is an opportunity to develop agonist ligand candidates with more potent bias properties. Following this concept, the researchers synthesized a series of derivatives based on apelin and MM07 that included M11 substitutions (alanine, phenylalanine, and a naphthalene ring). The results revealed that the derivative WN353, containing a larger naphthalene substituent, completely abolished the β-arrestin signal and exhibited strong G protein-biased characteristics.
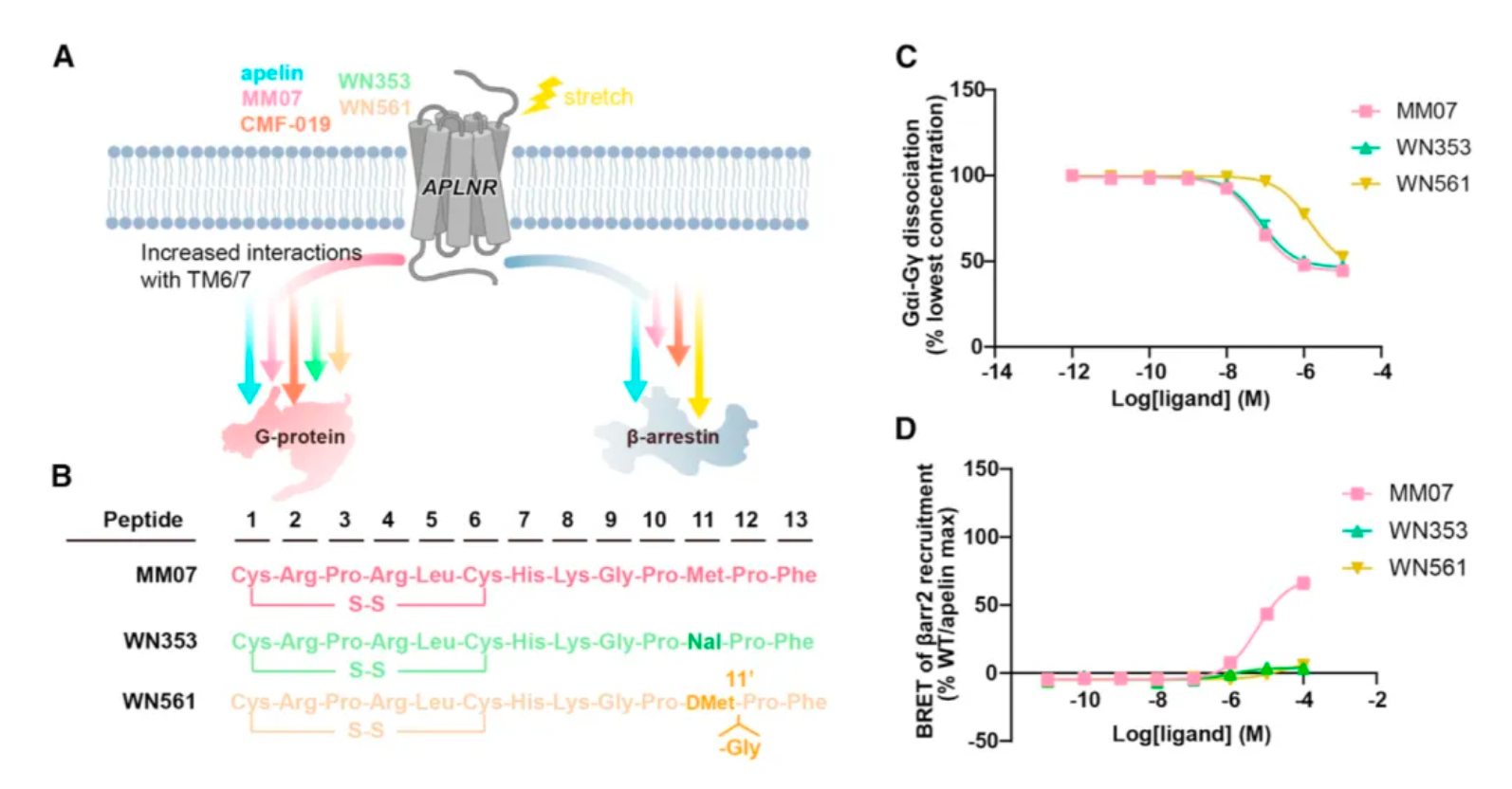
Researchers have engineered modifications to the structure of MM07, replacing methionine with D-methionine (denoted as m11), followed by the introduction of a glycine after m11, and adjusting the length of the peptide chain, thereby creating WN561. The results indicate that WN561 also acts as a pure G-protein-biased agonist at the APLNR.
Furthermore, the effects of WN561 in animal models were tested by the researchers. WN561 was effective both in vitro and in vivo in inhibiting cardiomyocyte hypertrophy, significantly reducing the heart weight/body weight ratio, cardiomyocyte area, and the expression of hypertrophic marker genes, as well as improving cardiac function indicators such as ejection fraction and short-axis shortening rate. Notably, after the removal of pathological stimuli, WN561 did not exacerbate myocardial hypertrophy during prolonged treatment, performing better than traditional APLNR agonists such as apelin, MM07, and CMF-019. These findings suggest that WN561 has superior cardioprotective effects and a lower hypertrophic side effect profile, making it an ideal candidate drug for the treatment of cardiovascular diseases, especially myocardial hypertrophy.
Reference:
https://www-cell-com.libproxy1.nus.edu.sg/cell/abstract/S0092-8674(24)00125-9



