Targeting FGFR in Cancer Therapy: From Biological Mechanisms to Clinical Breakthroughs
The fibroblast growth factor receptor (FGFR) family consists of transmembrane receptor tyrosine kinases that regulate a broad range of biological processes, including embryonic development, wound healing, and angiogenesis. Upon binding of FGFs (fibroblast growth factors) to their corresponding FGFRs, downstream signaling pathways—such as RAS-RAF-MAPK, PI3K-AKT/mTOR, STATs, and PLCγ/PKC—are activated, orchestrating cellular proliferation, survival, migration, and differentiation. In oncology, aberrant activation of FGFR signaling is considered a key oncogenic driver, with FGFR alterations identified in approximately 7% of human cancers. These alterations contribute to uncontrolled cell behavior and disease progression, making small-molecule FGFR inhibitors a hot area of research in targeted cancer therapy.
Biological Functions of FGFR Signaling
The FGFR family comprises four highly conserved transmembrane receptor tyrosine kinases—FGFR1 through FGFR4. Upon ligand binding with specific FGFs, these receptors form ternary complexes that initiate complex intracellular signaling cascades. FGFRs play crucial roles in morphogenesis during embryogenesis and are also involved in adult tissue repair, angiogenesis, and the maintenance of stem cell pluripotency, thus serving as a vital bridge between normal physiology and disease states.
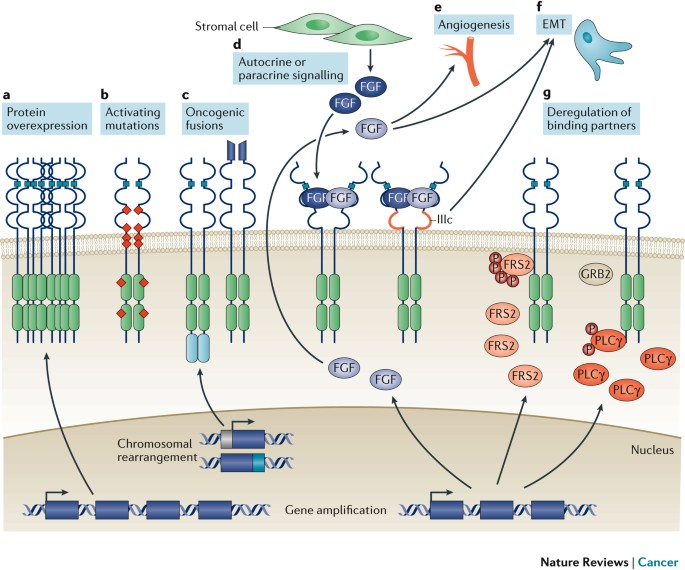
Upon FGF-FGFR interaction, multiple downstream signaling pathways are activated. These include:
·RAS-RAF-MAPK, primarily driving mitogenic signaling;
·PI3K-AKT/mTOR, which promotes cell survival and metabolic regulation;
·STATs, acting as transcription factors that modulate gene expression in response to extracellular stimuli and participate in immune and inflammatory responses;
·PLCγ/PKC, which regulate calcium signaling and cytoskeletal remodeling, essential for cell motility and morphological changes.
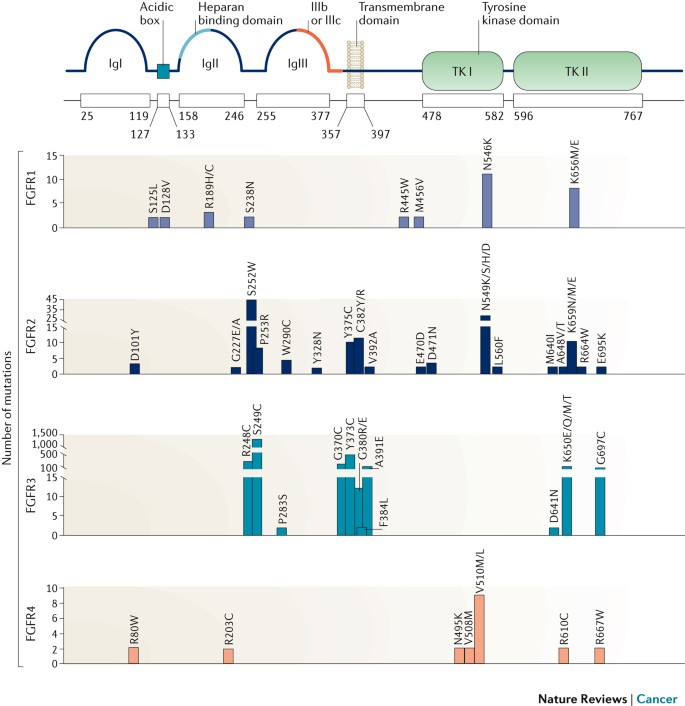
While FGFR signaling maintains homeostasis under physiological conditions, dysregulation due to genetic mutations or environmental stress can lead to pathological consequences. In cancer, FGFR pathway dysregulation is recognized as a major oncogenic mechanism. Alterations—including gene amplification, point mutations, and chromosomal rearrangements—are found in about 7% of human cancers. These result in elevated FGFR expression or constitutive activation, driving uncontrolled proliferation, apoptosis resistance, invasiveness, and metastatic potential. FGFR signaling is also implicated in promoting epithelial–mesenchymal transition (EMT), a process contributing to cancer cell migration and therapy resistance.
Therapeutic Strategies Targeting FGFR Pathways
Given its pivotal role in various cancers, the FGFR signaling pathway has become a prominent target for therapeutic intervention. Several selective FGFR tyrosine kinase inhibitors (TKIs) have progressed into clinical trials and demonstrated promising anti-tumor efficacy. For instance, Erdafitinib became the first FDA-approved FGFR inhibitor for the treatment of locally advanced or metastatic urothelial carcinoma harboring specific FGFR2 or FGFR3 mutations. A number of other selective FGFR inhibitors are also in various stages of development, aiming to provide more precise and tolerable alternatives to conventional chemotherapy.
Beyond direct inhibition, combination therapies targeting FGFR signaling alongside other pathways are under active investigation to enhance efficacy and delay resistance.
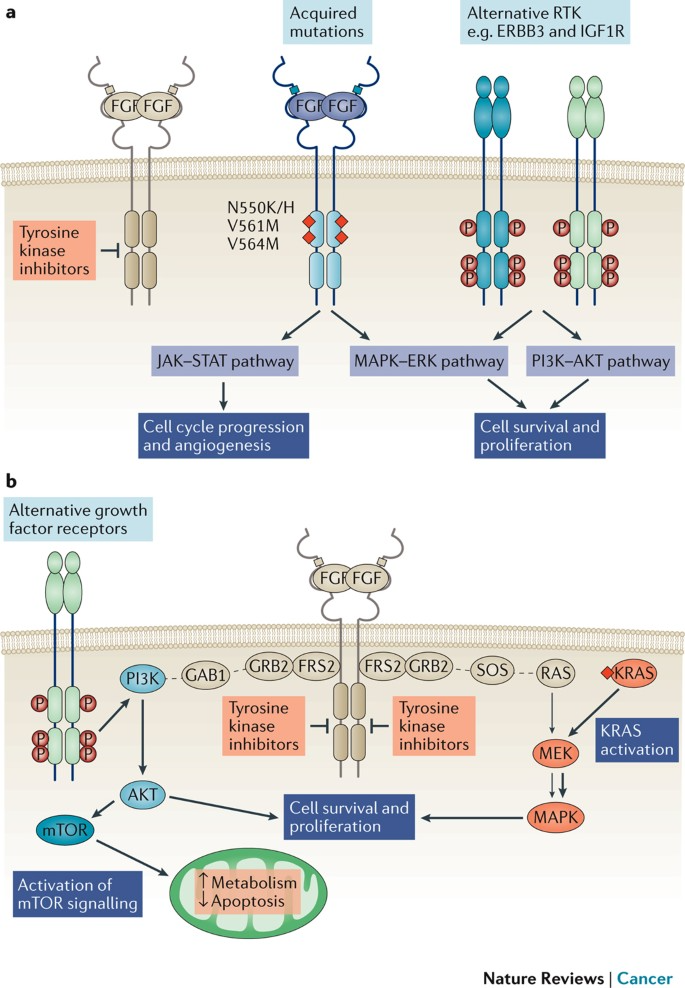
Mutations or overexpression of FGFR genes result in hyperactivation of the FGFR signaling cascade, promoting tumorigenesis. These genetic alterations—such as amplifications, point mutations, or rearrangements—lead to elevated FGFR protein levels or constitutive signaling activity, fueling malignant behaviors like proliferation, survival, invasion, metastasis, and therapy resistance.
According to an analysis of 4,853 solid tumor samples, FGFR alterations were identified in 7.1% of cases. Among these:
·Gene amplification was the most prevalent form (66%),
·Followed by point mutations (26%),
·And chromosomal rearrangements (8%).
FGFR1 aberrations were the most common, accounting for approximately 49% of all FGFR alterations, followed by FGFR3 (26%), FGFR2 (19%), and FGFR4 (7%). These findings underscore the relevance of FGFR-targeted therapies in precision oncology across multiple tumor types.
Research and Development Progress of FGFR Inhibitors
Given the critical role of the FGFR signaling pathway in tumorigenesis, scientists have been actively engaged in the development of FGFR inhibitors. These inhibitors are typically highly selective and specific, designed to precisely block the aberrant signaling mediated by FGFR while minimizing effects on non-target tissues. Currently, several selective FGFR inhibitors have entered clinical trials, and some have even received regulatory approval.
1. Erdafitinib
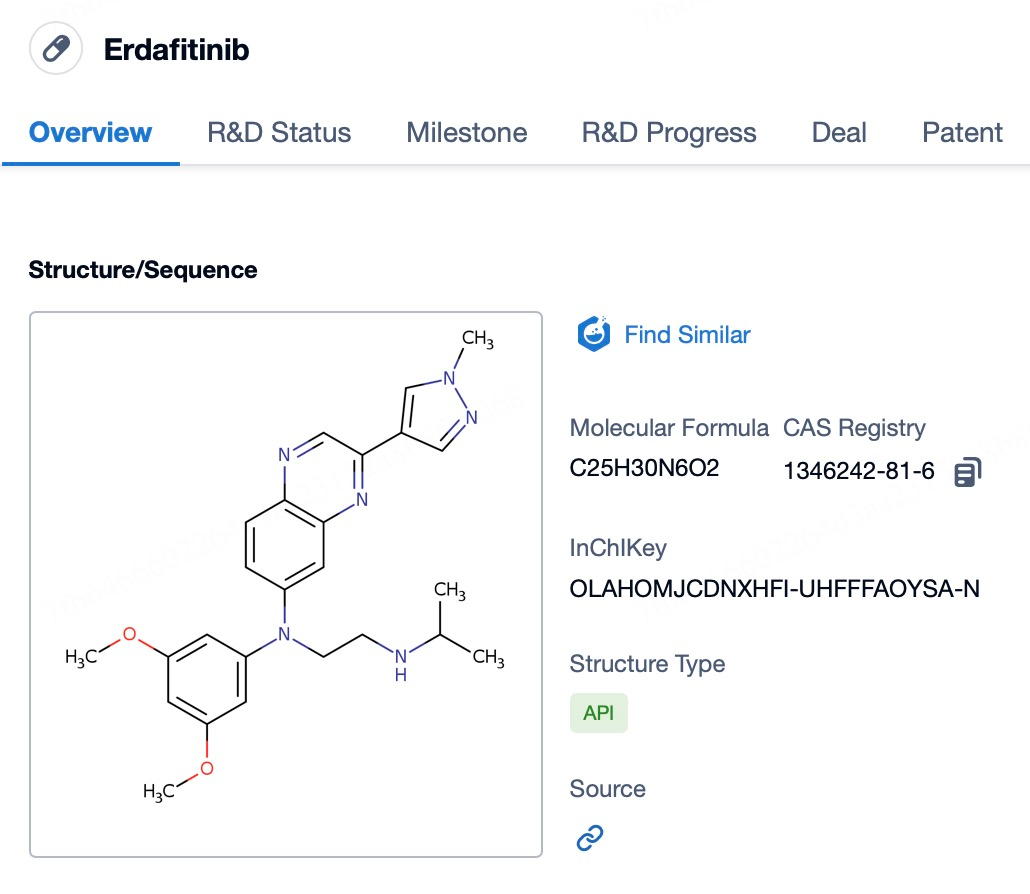
Erdafitinib (brand name Balversa) is the first selective pan-FGFR inhibitor approved by the U.S. Food and Drug Administration (FDA). It is specifically indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC) harboring specific FGFR3 or FGFR2 mutations. These patients often experience disease progression during or after platinum-containing chemotherapy, indicating poor response or acquired resistance to standard therapies. As an oral medication, erdafitinib effectively targets and inhibits the activity of FGFR1 through FGFR4, thereby counteracting abnormal cell proliferation and survival driven by dysregulated FGFR signaling.
The mechanism of action of erdafitinib involves its specific binding to the ATP-binding pocket of FGFR family kinases, thereby inhibiting the functions of these receptor tyrosine kinases. When the FGFR signaling pathway is aberrantly activated due to genetic mutations or other causes, it can promote tumor cell growth, survival, and migration. By blocking this abnormal signaling, erdafitinib helps to slow or even halt tumor progression, and it may induce tumor shrinkage. This mechanism makes erdafitinib an ideal treatment option for patients with specific FGFR alterations, offering a critical alternative for individuals who have failed to respond to conventional therapies.

Initially, erdafitinib received FDA accelerated approval in April 2019 based on the results of the Phase II BLC2001 clinical trial, which demonstrated significant efficacy in patients with FGFR3 or FGFR2 mutations. Subsequently, on January 19, 2024, the FDA granted full approval for erdafitinib in the treatment of adult patients with locally advanced or metastatic urothelial carcinoma harboring susceptible FGFR3 or FGFR2 alterations, following disease progression after platinum-containing chemotherapy.
In China, the National Medical Products Administration (NMPA) officially approved erdafitinib tablets, developed by Johnson & Johnson, on January 13, 2024. The indication is for adult patients with unresectable locally advanced or metastatic urothelial carcinoma carrying susceptible FGFR3 mutations who have experienced disease progression during or after prior therapy with at least one line of anti-PD-1 or anti-PD-L1 inhibitors.
The Phase III THOR (BLC3001) trial demonstrated erdafitinib's outstanding performance in overall survival (OS), progression-free survival (PFS), and objective response rate (ORR). Compared with chemotherapy, erdafitinib showed a statistically significant improvement in OS. The median OS for the erdafitinib group was 12.1 months versus 7.8 months in the chemotherapy group (HR=0.64; 95% CI, 0.47–0.88; p=0.0050), indicating that erdafitinib reduced the risk of death by 36% compared to chemotherapy. Furthermore, the median PFS in the erdafitinib group was 5.6 months, compared with 2.7 months in the chemotherapy group. The ORR with erdafitinib was also higher at 35.3%, compared with 8.5% in the chemotherapy group.
Despite the promising clinical benefits of erdafitinib, its use may be associated with certain adverse events. The most common adverse reactions (≥20%) include hyperphosphatemia, diarrhea, stomatitis, dry mouth, decreased appetite, dry skin, anemia, and constipation. To ensure safe and effective treatment, physicians should closely monitor patients' health and adjust the dosage or provide supportive care as necessary to manage side effects.
2. Pemigatinib
Pemigatinib (brand name: Pemazyre) is a highly selective FGFR1-3 inhibitor. It received accelerated approval from the U.S. Food and Drug Administration (FDA) for the treatment of pre-treated advanced cholangiocarcinoma patients with FGFR2 fusions or rearrangements. These patients often experience disease progression following first-line standard therapy and face significant unmet medical needs due to the lack of effective second-line or later therapeutic options. The advent of pemigatinib offers new hope for these patients.
Pemigatinib specifically binds to the ATP-binding pocket of FGFR, thereby inhibiting receptor tyrosine kinase autophosphorylation and downstream signaling pathways. This inhibits tumor cell growth and dissemination. This targeted therapeutic approach allows precise action on tumor cells with specific genetic alterations, reducing the impact on normal cells and improving treatment efficacy and safety.
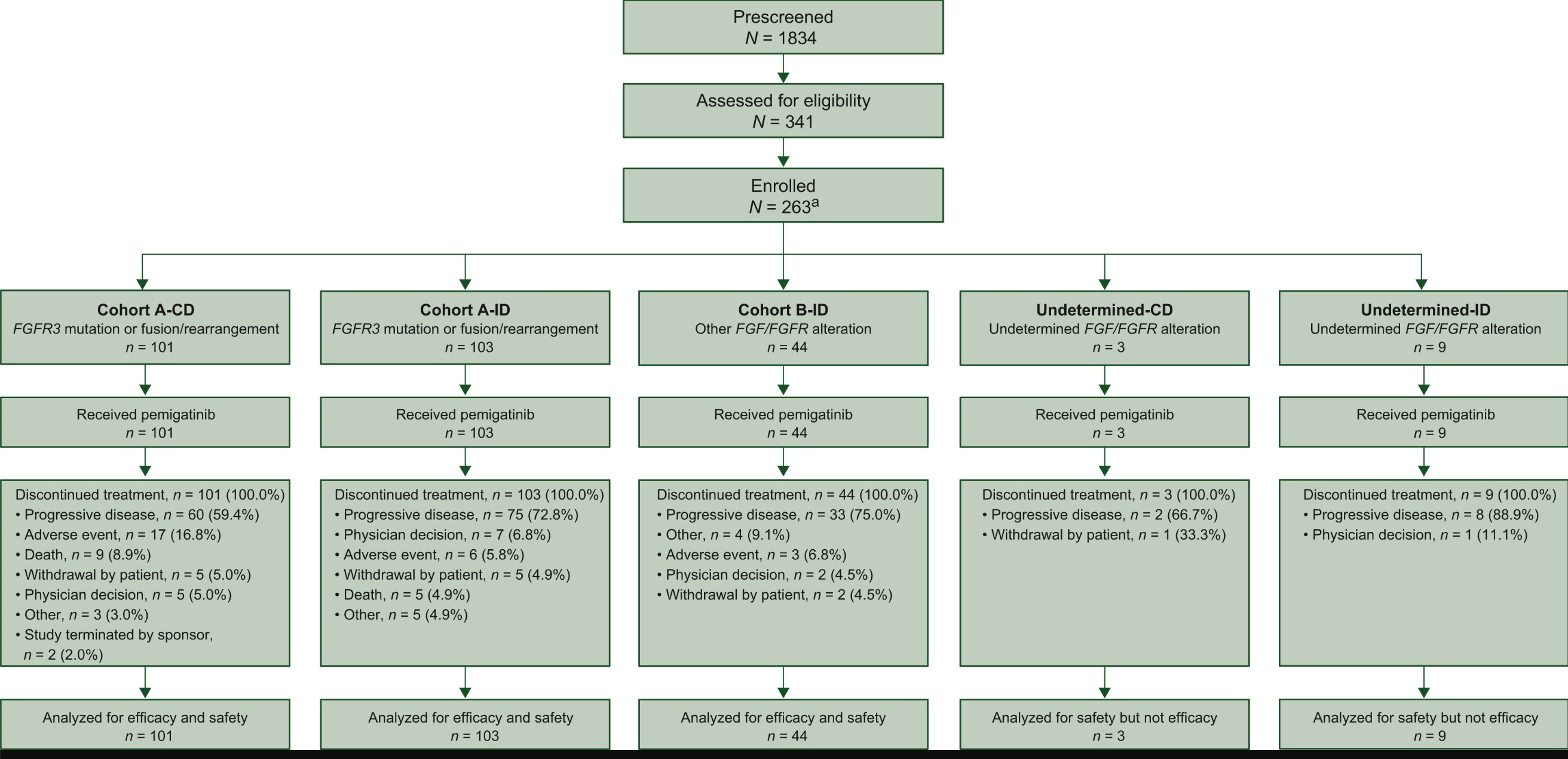
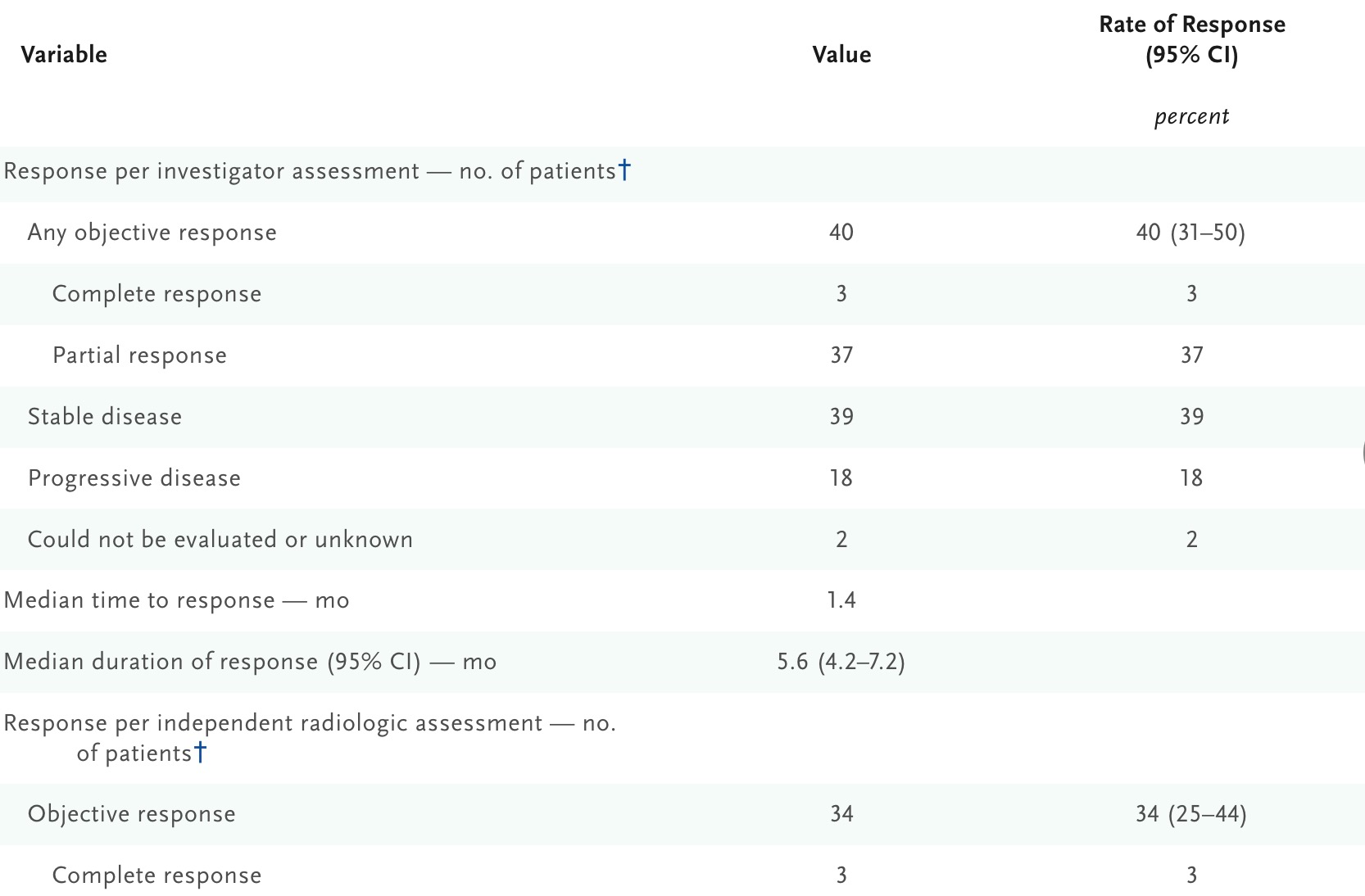
The approval of pemigatinib was based on data from the FIGHT-202 trial, a Phase II, single-arm, open-label clinical study in patients with locally advanced or metastatic cholangiocarcinoma harboring FGFR2 fusions or rearrangements, who had previously received systemic therapy and whose tumors were unresectable. In cohort A of the trial, which included patients with FGFR2 fusions or rearrangements, the median follow-up was 15 months. Pemigatinib monotherapy achieved an overall response rate (ORR) of 36%, with 4 cases of complete response (3.7%) and 36 cases of partial response (33.3%). The median duration of response (DoR) was 9.1 months. Furthermore, the study demonstrated good tolerability consistent with the mechanism of action of pemigatinib. Common adverse events included hyperphosphatemia (58.5%), alopecia, diarrhea, nail toxicity, and fatigue.
In terms of safety, pemigatinib exhibited relatively favorable tolerability. Approximately 68.7% of the participants experienced Grade 3 or higher adverse events, but most could be managed by dose adjustments or supportive care measures. The most common Grade 3 or higher adverse events included hypophosphatemia (14.3%) and other significant side effects such as arthralgia and oral mucositis (6.1% each), abdominal pain, fatigue, and hyponatremia (5.4% each). Palmar-plantar erythrodysesthesia syndrome and hypotension were less frequently observed. Notably, ocular toxicity represents a potential risk with the use of pemigatinib, necessitating regular ophthalmologic monitoring to detect or manage any eye-related issues.
In addition to its approval in the United States, pemigatinib has also been authorized for the treatment of FGFR2 fusion-positive unresectable biliary tract cancer (BTC) patients who have progressed after chemotherapy in Japan. In Europe, it is approved for adult patients with locally advanced or metastatic cholangiocarcinoma harboring FGFR2 fusions or rearrangements, whose disease has progressed following at least one prior systemic therapy. In China, Innovent Biologics has submitted a new drug application for pemigatinib, and this has been accepted by the National Medical Products Administration (NMPA), signaling an active evaluation of the drug's suitability for Chinese patients.
3. Other Selective FGFR Inhibitors Under Development
In addition to the two aforementioned selective FGFR inhibitors that have been approved, several other small-molecule compounds are undergoing clinical development at various stages. For instance, BGJ398 is a highly selective tyrosine kinase inhibitor targeting FGFR1 that has demonstrated effective inhibition of acute myeloid leukemia cell lines in vitro. Similarly, Irpagratinib (ABSK011), an orally active FGFR4 inhibitor, has been shown to effectively suppress tumor growth in FGFR4-driven liver cancer models in vivo. The development of these new agents not only broadens the application scope of FGFR-targeted therapies but also provides potential solutions to address emerging resistance issues.
Conclusion
In summary, the FGFR signaling pathway is not only critical for normal physiological processes but also acts as a driver for the development of many cancers when dysregulated. By specifically blocking abnormal FGFR-mediated signaling cascades, next-generation selective FGFR inhibitors such as erdafitinib and pemigatinib have demonstrated remarkable efficacy in clinical trials and received regulatory approval for the treatment of specific types of advanced cancers. These drugs not only offer new therapeutic options for patients harboring specific FGFR alterations but also advance the field of precision medicine. With further accumulation of clinical data and technological progress, FGFR-targeted therapies are expected to expand their application scope in the future, improving patient outcomes and quality of life. Moreover, ongoing research will help address potential resistance issues, ensuring sustained therapeutic efficacy over the long term.
How to obtain the latest research advancements in the field of biopharmaceuticals?
In the Synapse database, you can keep abreast of the latest research and development advances in drugs, targets, indications, organizations, etc., anywhere and anytime, on a daily or weekly basis. Click on the image below to embark on a brand new journey of drug discovery!
Reference
- 1.Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017 May;17(5):318-332. doi: 10.1038/nrc.2017.8. Epub 2017 Mar 17. PMID: 28303906.
- 2.Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, Fleming M, Rezazadeh A, Mellado B, Varlamov S, Joshi M, Duran I, Tagawa ST, Zakharia Y, Zhong B, Stuyckens K, Santiago-Walker A, De Porre P, O'Hagan A, Avadhani A, Siefker-Radtke AO; BLC2001 Study Group. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N Engl J Med. 2019 Jul 25;381(4):338-348. doi: 10.1056/NEJMoa1817323. PMID: 31340094.
- 3.Necchi A, Pouessel D, Leibowitz R, Gupta S, Fléchon A, García-Donas J, Bilen MA, Debruyne PR, Milowsky MI, Friedlander T, Maio M, Gilmartin A, Li X, Veronese ML, Loriot Y. Pemigatinib for metastatic or surgically unresectable urothelial carcinoma with FGF/FGFR genomic alterations: final results from FIGHT-201. Ann Oncol. 2024 Feb;35(2):200-210. doi: 10.1016/j.annonc.2023.10.794. Epub 2023 Nov 11. PMID: 37956738.



