Progress in the Research of Therapeutic Peptides Targeting GPCRs
GPCRs, as key signal transduction molecules on the cell surface, regulate various physiological processes and constitute one-third of the targets for clinical drug applications. Based on structural similarities, GPCRs are divided into several families, including A, B, C, etc. Although most drugs targeting GPCRs are small molecules, many GPCRs have endogenous ligands that are peptides containing fewer than 50 amino acids. According to the pharmacology guidelines of the International Union of Basic and Clinical Pharmacology (IUPHAR), about one-third (64 types) of class A GPCRs bind to endogenous peptides; 20 types of class B GPCRs are activated by 15 endogenous peptides, and these receptors are classified according to the ligands they recognize into families such as calcitonin, corticotropin-releasing factor, glucagon, parathyroid hormone (although it has 84 amino acids, it is still typically considered a peptide), vasoactive intestinal peptide (VIP), or pituitary adenylate cyclase-activating peptide (PACAP), etc.
Typically, endogenous peptides can bind to GPCRs on the cell surface, bridging paracrine and autocrine signaling and regulating the downstream signaling pathways of GPCRs. Peptides are one of the largest and oldest categories of intercellular chemical messengers. The pioneering development of using these naturally occurring peptides as therapeutic drugs dates back to the application of insulin in the 1920s.
Insulin primarily acts on tyrosine kinase receptors, and the development of this therapy has made full use of the outstanding pharmacokinetic properties of peptides: high affinity, selectivity, and potency. Consistent with the observed effects of insulin, most other peptide therapies are well-tolerated with fewer side effects. However, naturally occurring peptides are generally not suitable as effective therapeutic drugs. The development of peptide drugs is limited by factors such as poor pharmacokinetic properties (short half-life, rapid degradation, high clearance), as well as insufficient oral bioavailability due to low intestinal stability and poor permeability. Therefore, to make most peptides effective drugs, strategies that address these issues need to be developed.
Peptide-based therapeutic drugs are experiencing a resurgence. Emerging innovative strategies include extending the half-life, stabilizing peptide chains, and enhancing resistance to proteolytic degradation, all of which significantly improve pharmacokinetic properties and oral bioavailability. A prime example of these strategies might be the development of glucagon-like peptide-1 (GLP-1) receptor agonists, which possess enhanced resistance to proteolytic degradation and reduced renal clearance. Numerous successful market products and a large array of novel approaches in preclinical stages (such as peptide stabilization, cyclization, and glycosylation) stem from these efforts. This success has also spurred the development of multifunctional peptides that combine agonist activities against two or more GPCRs within the same peptide, based on relatively high sequence homology and similar binding sites. Complementary pharmacokinetic strategies are expanding the variety of drugs acting on specific GPCRs.
Approved peptide therapeutic drugs
Most of the peptide drugs targeting GPCRs that have been approved for clinical use or are under development function as agonists, used to substitute for or enhance the low levels of endogenous peptides. The following figure summarizes peptide drugs targeting GPCRs.
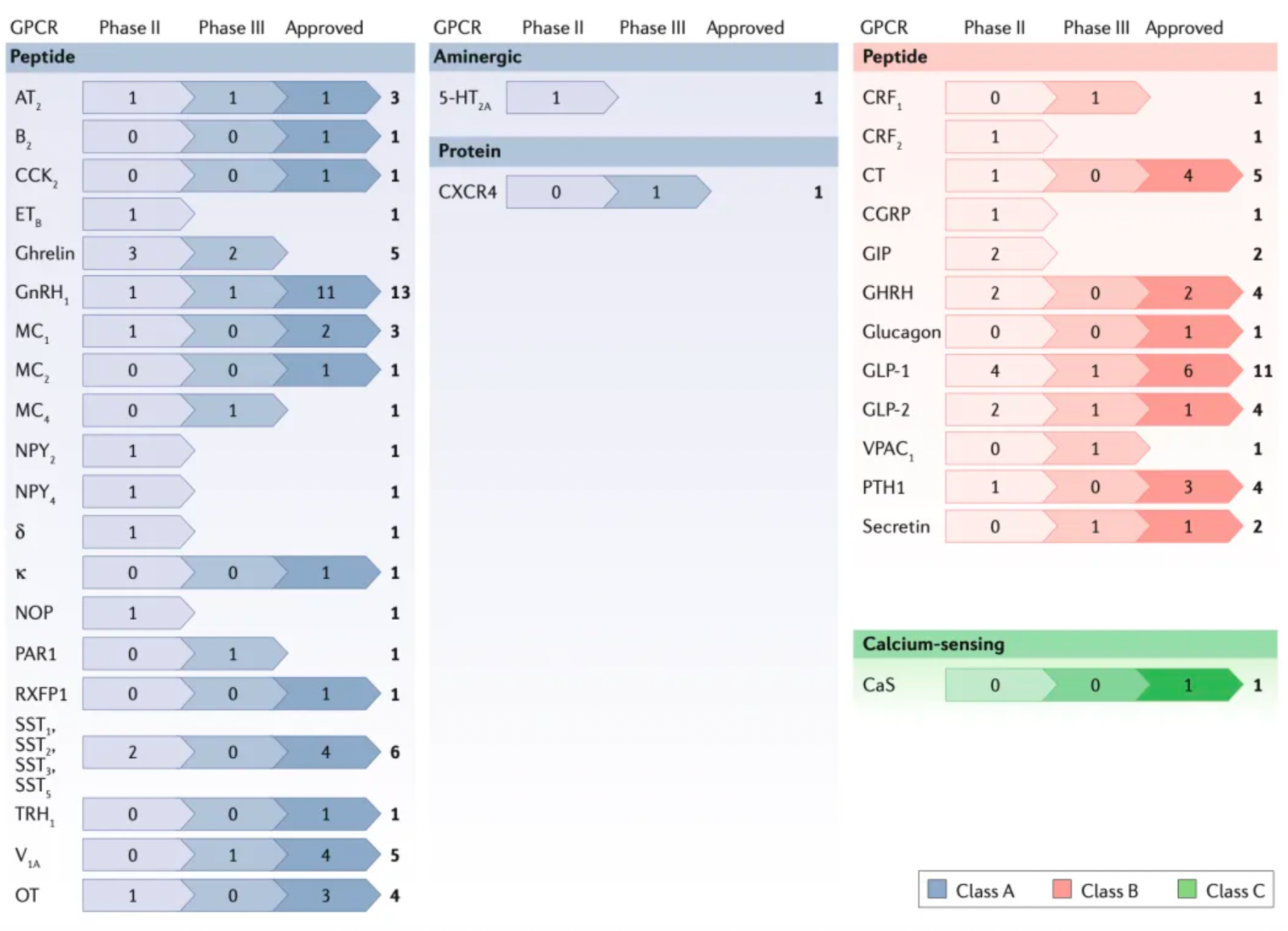
Within class A GPCRs, representative receptor targets include the μ and κ opioid receptors (abbreviated as MOR and KOR, respectively) for pain relief; oxytocin and vasopressin receptors for inducing labor; as well as apelin and angiotensin receptors for the treatment of cardiovascular diseases; and somatostatin receptors for treating acromegaly and Cushing's disease, among others.
Peptide therapies targeting Class B receptors include GLP-1 receptor agonists for the treatment of Type 2 Diabetes (T2D), GLP-2 receptor agonists for the treatment of Short Bowel Syndrome, as well as other synthetic or modified peptide hormones.
Currently, a few peptide antagonists have entered clinical trials, most of which target Class A GPCRs. For example, antagonists that block the action of Gonadotropin-Releasing Hormone (such as degarelix and abarelix) are used for cancer treatment.
Synthesis of Endogenous Peptide Analogs
The marketed synthetic endogenous peptide drugs targeting Class A and Class B GPCRs include those targeting the angiotensin receptor, melanocortin receptor, thyrotropin-releasing hormone receptor, vasopressin receptor, oxytocin receptor, calcitonin receptor, glucagon receptor, parathyroid hormone receptor, among others.
According to statistics, peptide drugs targeting Class A GPCRs are mainly high-affinity (pKi=8.4) and high-efficacy (pEC50=8.5) agonists. However, they typically exhibit a very short plasma half-life following intravenous administration, with a median value of about 5.3 minutes. This reflects their rapid degradation by peptidases or their higher rate of excretion, particularly through the kidneys. Theoretically, the levels of peptide drugs are expected to decrease to less than 1% of the original dose within 6 half-lives, which is approximately 32 minutes. Lastly, endogenous peptides usually have a low rate of binding to plasma proteins, ensuring that most peptides in circulation are unbound and can directly interact with and activate the corresponding receptors. However, unbound peptides are also highly susceptible to renal clearance and cleavage by serum or tissue proteases, both of which can reduce the plasma half-life.
For all Class B GPCRs, the pharmacokinetic and pharmacodynamic median values of their endogenous ligands are similar to those of Class A receptor peptides: high efficacy (pEC50=9.9), accompanied by a short plasma half-life (15 minutes) and a low volume of distribution (approximately 9.9 L).

It is worth mentioning that the short plasma half-lives of at least two classes of peptides have been utilized for clinical benefit. The peptide drug with the shortest plasma half-life is Angiotensin II. Synthetic analogs of this Angiotensin II are used to treat critically ill patients with distributive shock due to sepsis, where abnormal distribution of blood to the smallest vessels leads to insufficient systemic blood supply, potentially resulting in death. This drug binds to the AT1 receptors on vascular smooth muscle, and after intravenous administration, it effectively raises blood pressure within a median time of 5 minutes. Its short half-life (less than 1 minute) ensures that the possibility of hypertensive crisis due to overdose is minimal. Similarly, oxytocin, which was sequenced and synthesized in the 1950s, is used to induce labor and immediately enhance contractions upon intravenous administration, but its effects wear off within an hour, reflecting its short half-life of several minutes.
Modified peptides
Taking Semaglutide as an example, modifications and redesigns have significantly extended its half-life (approximately 168 hours). This extension is achieved by using a fatty acid linker, which allows the molecule to form non-covalent reversible interactions with albumin, thereby reducing renal excretion. Simultaneously, alpha-aminobutyric acid has been introduced at position 8 of Semaglutide to reduce metabolism mediated by Dipeptidyl Peptidase-4 (DPP-4). Additionally, Albiglutide and Dulaglutide utilize different protein linking strategies. These two GLP-1 receptor agonists are covalently linked to large molecules (albumin and the Fc fragment of human IgG4, respectively). In Albiglutide, the GLP-1 sequence at position 8 is also modified by substituting glycine with alanine near the DPP-4 cleavage site to reduce metabolism.
Another exemplary case is Lutathera ([177Lu]-dotatate) used for the treatment of neuroendocrine tumors, representing a recent example of peptide receptor radionuclide therapy. In this compound, the peptide dotatate, an agonist of the somatostatin receptor, is labeled with lutetium [177]. After the peptide binds to somatostatin receptors highly expressed on tumor surfaces, the radiation emitted causes tumor cell death, while the limited range of the particles minimally affects the surrounding healthy tissue due to their short penetration depth.
Peptide Design Optimization Strategies
Recent approvals of peptide drugs have shown trends such as high sustained target efficacy combined with increased plasma half-lives, reduced enzymatic metabolism, and decreased renal clearance. For example, degarelix is a synthetic derivative of GnRH that blocks the binding of endogenous peptides to pituitary receptors and is used to treat prostate cancer. After subcutaneous injection, degarelix forms a drug depot at the injection site, with the drug slowly releasing into the bloodstream, producing a plasma half-life of 42-70 days. Clearance is unaffected, primarily through hepatic-biliary hydrolysis and renal excretion of unchanged drug. Additionally, lanreotide is a somatostatin agonist that functions mainly by binding to SST2 and SST5 receptors, used to inhibit growth hormone release to treat acromegaly; it also forms a drug depot at the injection site with a 22-day plasma half-life and includes non-natural amino acids.
Peptides, being charged and hydrophilic molecules, typically lack oral bioavailability. Decades after the synthetic modification of peptides, most peptide drugs still possess the intrinsic drawback of low membrane permeability, which limits their oral bioavailability and tissue distribution, including distribution to the CNS. Emisphere's use of a pharmacologically inactive small molecule enhancer, N-[8-(2-hydroxybenzoyl)amino] caprylate (SNAC), co-formulated with Semaglutide, results in the oral drug semaglutide having increased transcellular permeability and about 4% bioavailability.
Experimental methods to enhance brain permeability include linking the drug neurotensin to the brain barrier-penetrating peptide angiopep-2, thus increasing the blood-brain barrier crossing capability by approximately tenfold through receptor-mediated transcytosis. This approach is sufficient to reverse pain behaviors in animal models of neuropathic and bone cancer pain.
Cyclization helps in resistance to metabolism, and its hydrophobicity enhances absorption by intestinal cells. Despite very low bioavailability via the oral route (0.08%-0.16%), this is sufficient to achieve clinically effective plasma concentrations. Desmopressin is one example of an orally administered peptide drug, showcasing the potential of engineering peptides to possess oral activity.
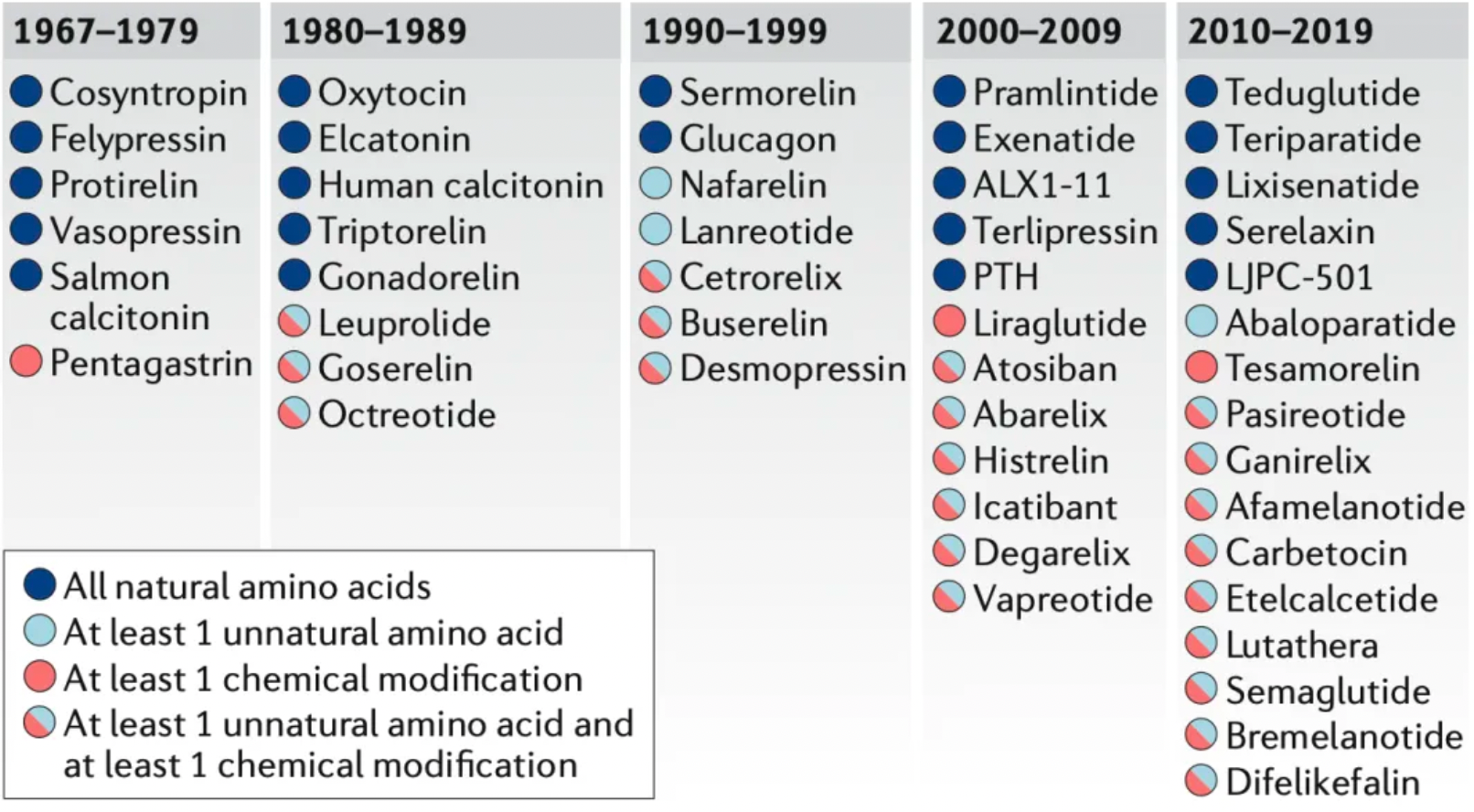
The new generation of peptide drugs often incorporates unnatural amino acid residues, such as D-amino acids, N-methyl amino acids, or residues with unnatural side chains. Chemical modifications—non-amino acid monomers added to peptides during synthesis—are also becoming increasingly important. These modifications include N-terminal modifications, C-terminal capping, fatty acids, and polyethylene glycol (PEG) groups. Among 49 peptide drugs approved for targeting GPCRs studied by researchers, 22 contain only natural amino acids, 3 contain at least one unnatural amino acid, 3 have at least one chemical modification, and 21 contain both chemical modifications and unnatural amino acids.
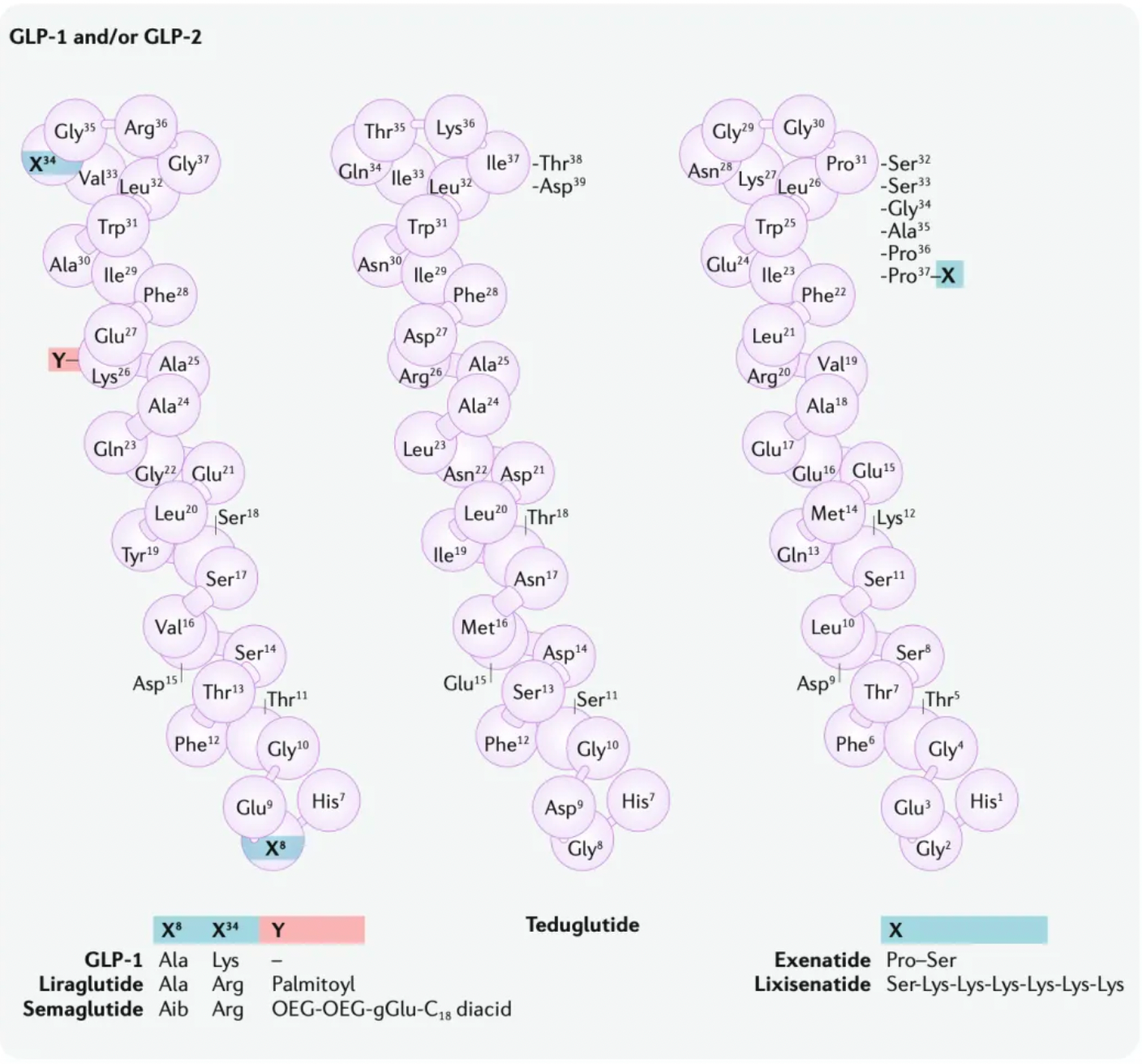
Classic Case: Development of GLP-1 Agonist Peptides
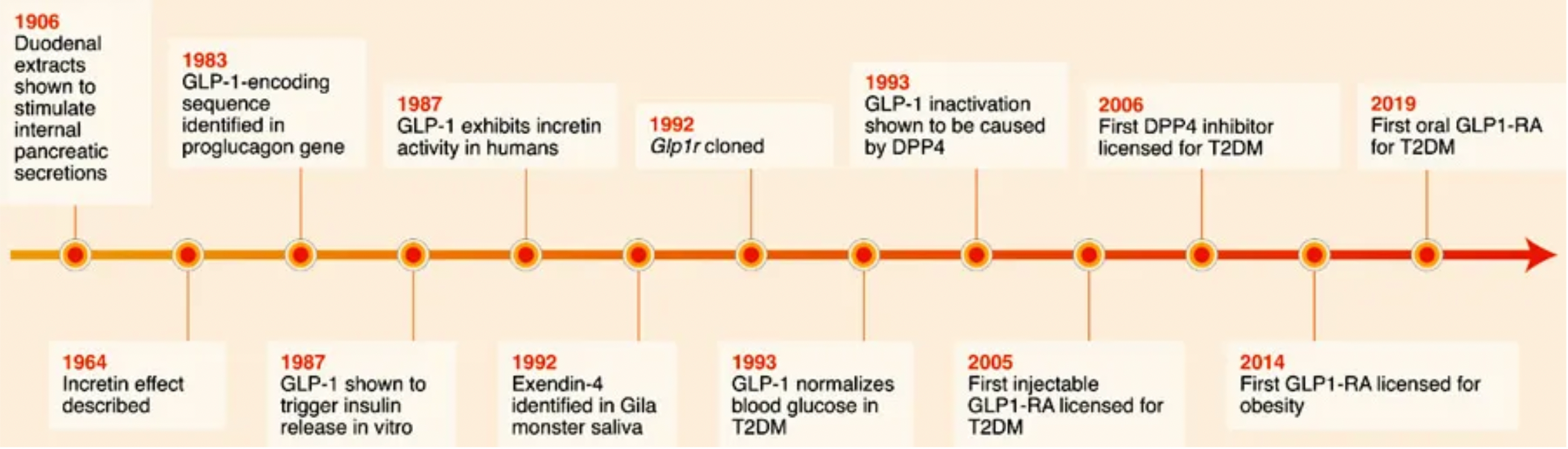
Exenatide is the first clinically-approved GLP-1 receptor agonist. In 1992, scientists identified a novel peptide hormone, exendin-4, from the saliva of the Heloderma suspectum. Exendin-4 shares many pharmacological properties with GLP-1: enhancing insulin secretion and reducing plasma glucose levels. However, unlike GLP-1, exendin-4 is resistant to enzymatic cleavage and inactivation by DPP4. As exenatide, it was approved in 2005 as an adjunct therapy for Type 2 Diabetes (T2D) and as a monotherapy in 2009. Although exenatide is widely used in the treatment of T2D, its short plasma half-life requires frequent (twice daily) subcutaneous injections, which limits its efficacy, leads to poor patient compliance, and increases the risk of additional side effects such as injection site infections.
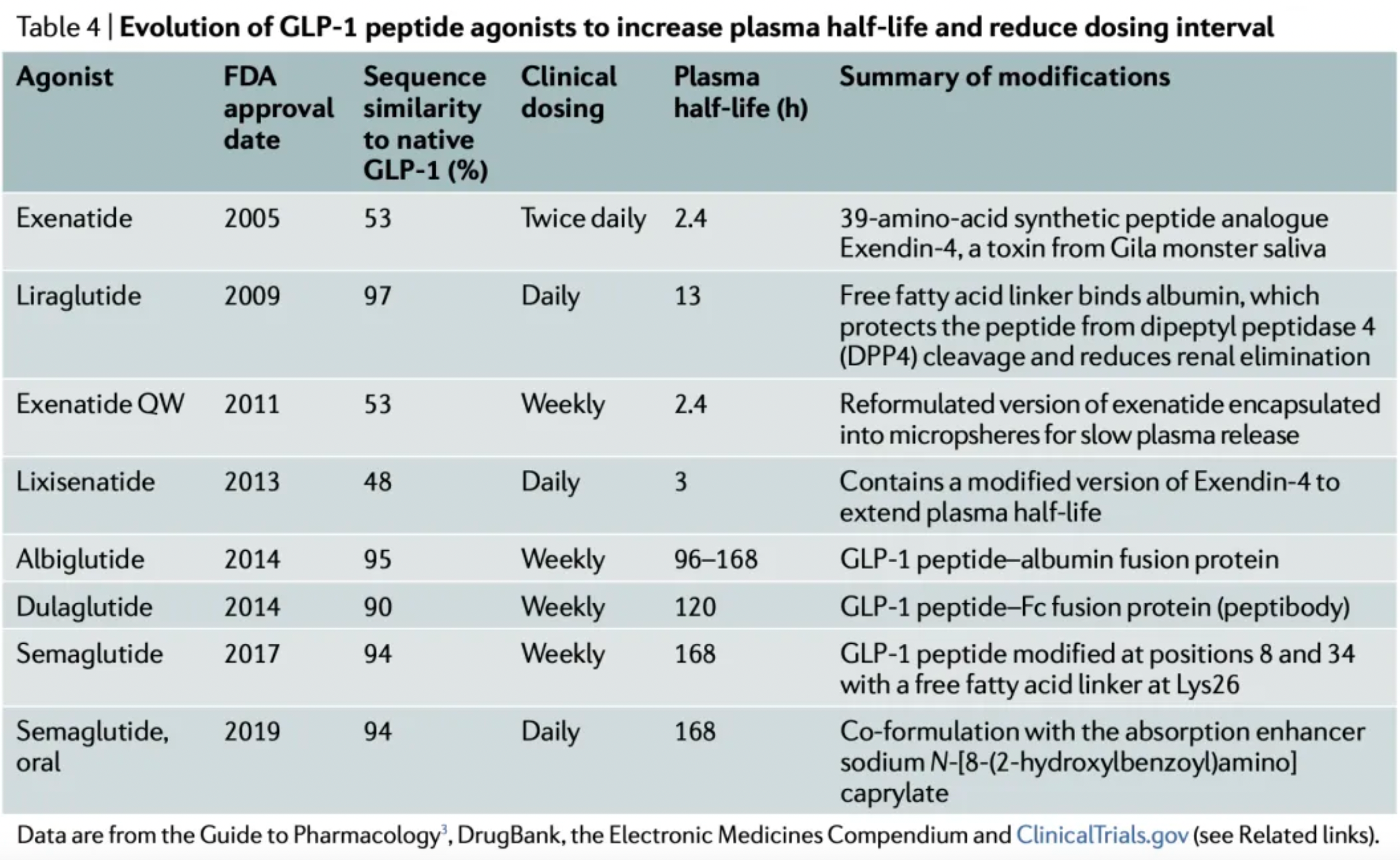
Following the success of exenatide, researchers have adopted multiple strategies to increase the plasma stability and half-life of GLP-1 receptor agonists, to reduce renal clearance, and to enhance oral bioavailability.
These strategies are generally divided into two approaches. One approach focuses on extending the plasma half-life of exenatide, which led to the development of once-weekly Exenatide (QW) and lixisenatide. Exenatide QW involves reformulating exenatide into microspheres composed of the biodegradable polymer poly(D,L-lactide-co-glycolide), thus extending the dosing interval to once weekly. In lixisenatide, modifications to the exenatide sequence, including the addition of a lysine tail at the C-terminus, confer resistance to DPP4 enzyme cleavage and prolong the plasma half-life.
The second strategy focuses on modifying the natural GLP-1 peptide, resulting in the production of liraglutide, albiglutide, dulaglutide, and the recently approved semaglutide. Liraglutide, approved in 2009, is a human GLP-1 receptor agonist based on the natural GLP-1 peptide, with an amino acid substitution (Lys34Arg) and a C16 palmitic acid side chain attached via a glutamyl spacer at position 26. These modifications increase its binding to serum albumin, significantly reducing renal clearance and DPP4 enzyme cleavage. The modified GLP-1 is released from albumin at a slow, steady rate, resulting in slower degradation and reduced renal clearance compared to the mature endogenous form of GLP-1, GLP-17-37. Semaglutide, approved in 2017, incorporates the non-natural amino acid α-aminoisobutyric acid at position 8 to reduce DPP4 enzyme cleavage, and contains a substituted C18 dicarboxylic fatty acid at Lys26 that provides strong albumin binding. Both albiglutide and dulaglutide are peptide fusion protein variants. Each carries protection against DPP4 enzyme cleavage due to a substitution at position 8, and both include two GLP-1 molecules, either fused to human serum albumin (albiglutide) or covalently linked via a small peptide connector to the human IgG4-Fc heavy chain (dulaglutide), to increase the molecule's half-life through recycling and/or interaction with the neonatal Fc receptor (FcRn) or by increasing molecular weight.
Reference:
https://www-nature-com.libproxy1.nus.edu.sg/articles/s41573-020-0062-z



