Sequence-based drug design with AI: A Paradigm Shift in Drug Discovery
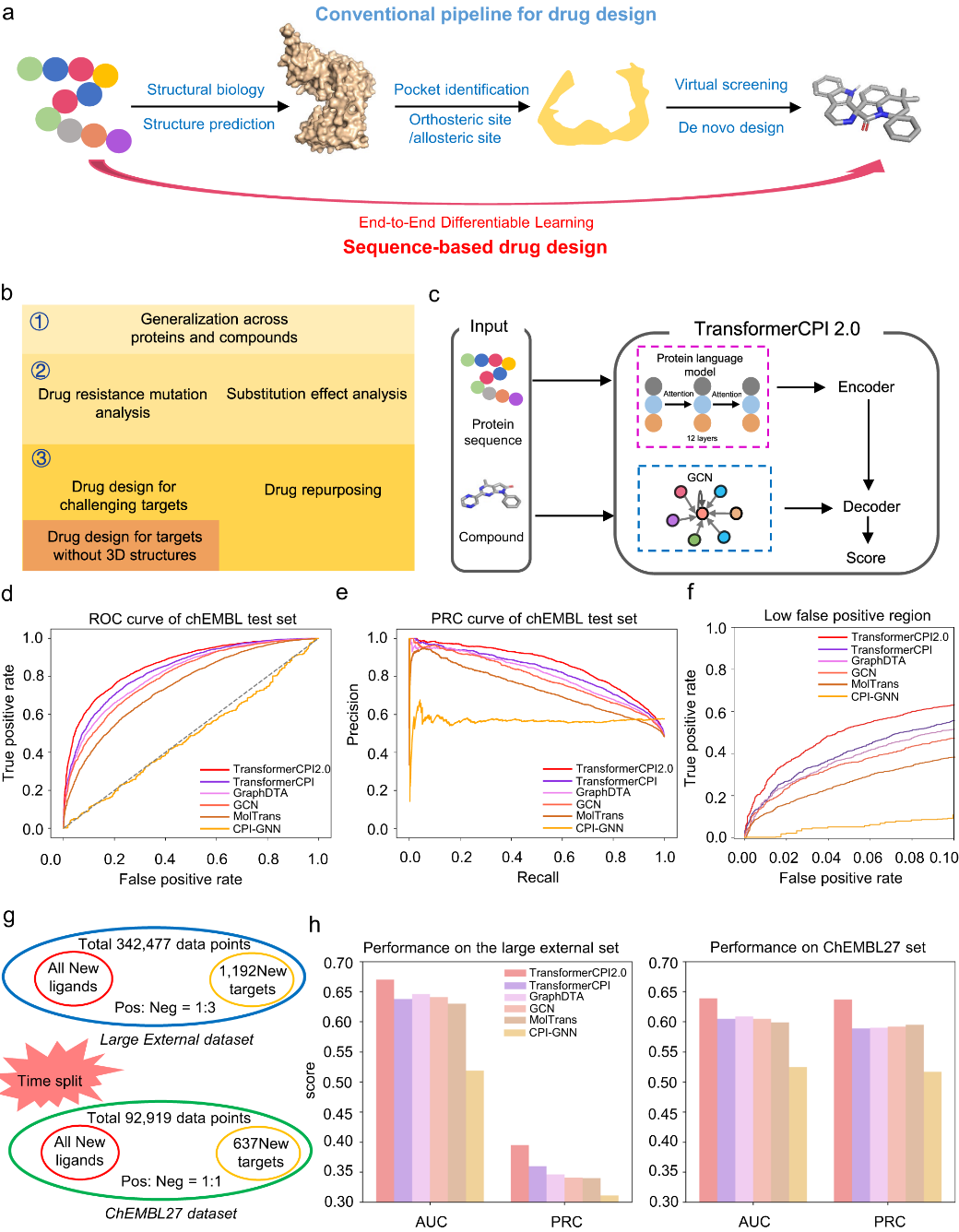
A team of scientists from Shanghai Institute of Materia Medica, Chinese Academy of Sciences, has proposed a novel approach to drug design, aiming to circumvent the intricate pipeline of protein structure-based drug development. The traditional method commences with the protein sequence, constructs a three-dimensional (3D) structure through structural biology or structure prediction, pinpoints binding pockets, and finally uncovers modulators via virtual screening or de novo design. However, this multi-step procedure has inherent limitations, as each part requires independent optimization. These limitations include the absence of high-resolution structures for numerous proteins, the challenge of accurately predicting active sites, defining binding pockets for novel targets with multiple domains, and anticipating allosteric sites.
Leveraging the power of AI
To address these issues, the researchers introduced the sequence-to-drug concept, a method that uncovers modulators straight from protein sequences, eliminating intermediary steps. This concept employs end-to-end differentiable learning, executing the entire learning process in a self-consistent, data-efficient manner, potentially mitigating the error accumulation inherent to complex pipelines.
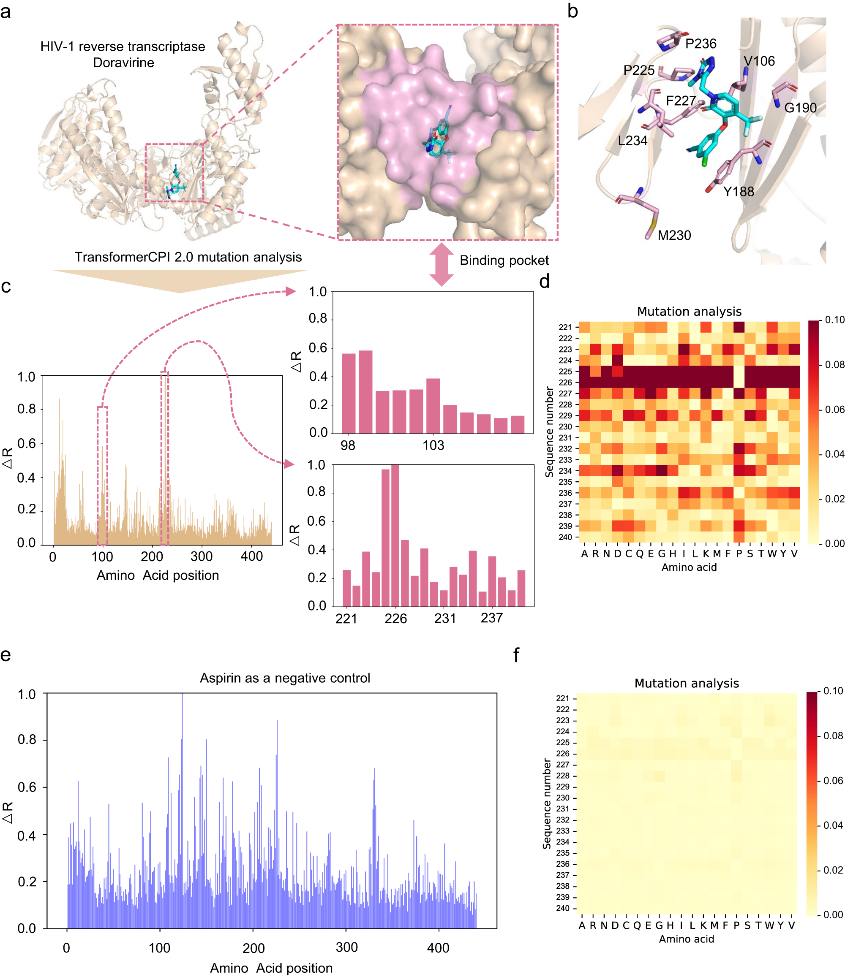
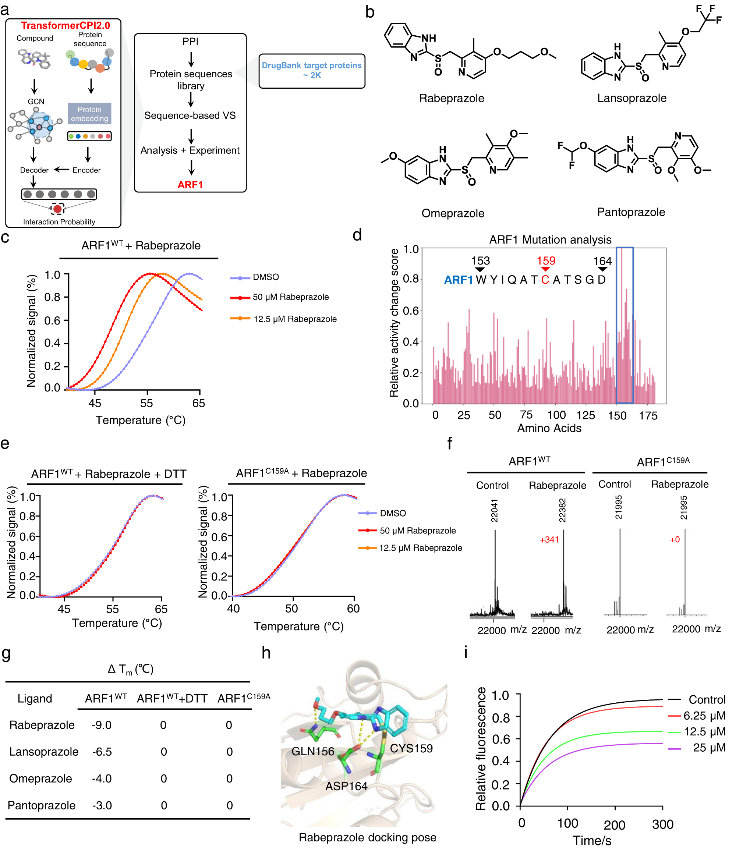
The team designed the TransformerCPI2.0 tool to validate this concept, demonstrating its general applicability across proteins and chemical space. By conducting case studies, the model's learning efficiency was evaluated, and its acquisition of knowledge was confirmed to be in line with expectations, rather than merely showcasing data bias. TransformerCPI2.0 was employed to explore novel potential treatments for challenging targets, namely speckle-type POZ protein (SPOP) and ring finger protein 130 (RNF130), both of which lack existing 3D structures. Furthermore, a potential new target for proton pump inhibitors (PPIs), ADP-ribosylation factor 1 (ARF1), was identified by the researchers.
New hits for challenging drug targets
SPOP is an E3 ubiquitin ligase that plays a critical role in mediating the ubiquitination and degradation of substrate proteins. In ccRCC cells, SPOP is overexpressed and mislocalized to the cytoplasm, leading to decreased levels of its substrates PTEN and DUSP7. This, in turn, causes increased cell proliferation and tumor growth.
PTEN and DUSP7 are negative regulators of the oncogenic AKT and ERK signaling pathways. By mediating their degradation, cytoplasmic SPOP in ccRCC cells causes hyperactivation of AKT and ERK, driving cancer progression. Although SPOP is a promising drug target, effective chemical inhibitors have not yet been developed due to challenges with targeting protein-protein interactions.
But now, with the help of TransformerCPI2.0, new breakthroughs are made possible. By virtually screening compounds against SPOP, the researchers identified a top hit - 221C7. This compound contains a distinctive β-lactam ring not seen in prior SPOP inhibitors.
221C7 was experimentally validated to directly bind to SPOP's MATH domain and disrupt its interactions with substrates like PTEN/DUSP7. An optimized analog 230D7 exhibited anti-cancer effects in ccRCC models.
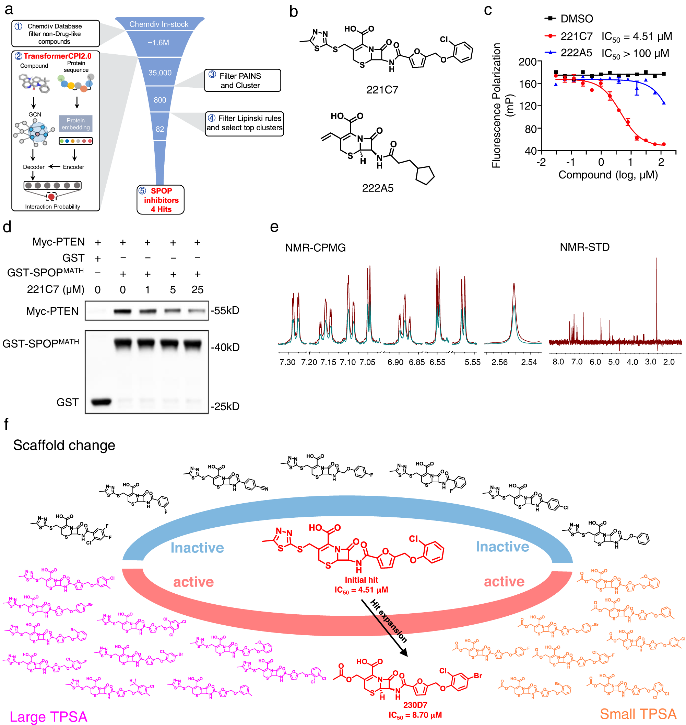
This success with SPOP demonstrates TransformerCPI2.0's ability to discover new inhibitor chemotypes by virtual screening against novel targets, using only sequence information. The researchers further applied TransformerCPI2.0 to find hits against RNF130, an E3 ligase lacking structural data and known binders. A compound iRNF130-63 was discovered and confirmed to directly bind RNF130.
New target for an existing drug
In addition, TransformerCPI2.0 could also be used to screen drugs against the entire human proteome, to identify novel protein targets for repurposing applications. Four PPIs were screened against 2,204 proteins in the DrugBank database, and the top scoring interaction was found to be with ARF1.
ARF1 is a small G protein involved in cancer stem cell lipid metabolism. Experimental validation showed that the PPIs directly bound and destabilized ARF1, with rabeprazole exhibiting the strongest effects. The binding occurred covalently at cysteine 159 of ARF1, and rabeprazole suppressed ARF1's GTP nucleotide exchange activity, inhibiting its function.
In colon cancer models, rabeprazole reduced ARF1 activity levels, causing lipid droplet accumulation in cancer stem cells and stimulating an anti-tumor immune response. Knockdown of ARF1 prevented rabeprazole's effects on tumor growth and immune activation, confirming that the mechanism is ARF1-dependent.
This study demonstrates the potential of TransformerCPI2.0 to identify novel drug-target interactions to enable repurposing applications, as shown by the discovery of PPIs as ARF1 inhibitors. Targeting ARF1 with PPIs represents a promising new approach to induce anti-cancer immunity. Further evaluation is still needed to assess their potential as ARF1-targeted immunotherapies.
Compared to conventional structure-based drug design, the novel approach is attractive because it conducts the entire learning process in a self-consistent and data-efficient manner, potentially avoiding the accumulation of errors in complex pipelines.
The proof-of-concept study illustrates that the sequence-to-drug concept provides a promising direction for rational drug design, especially for proteins lacking high-quality 3D structures. The new approach has the potential to enable the discovery of novel treatments for diseases with poorly defined protein targets.
Reference
Chen, L., Fan, Z., Chang, J. et al. Sequence-based drug design as a concept in computational drug design. Nat Commun 14, 4217 (2023).



