Tegileridine vs Oliceridine: Comparison of New Analgesic Drugs Targeting Opioid Receptors
In January, Jiangsu Hengrui Pharmaceuticals' Tegileridine Fumarate injection was approved for marketing in China, indicated for the treatment of moderate to severe postoperative pain following abdominal surgery. It is reported that this product is the first domestically developed Class 1 opioid analgesic innovation drug in China, offering a new treatment option for patients suffering from postoperative pain.
Tegileridine (SHR-8554) is a Class 1 new drug independently researched and developed by Hengrui Pharmaceuticals. It is a small molecule drug that targets the μ-opioid receptor (MOR), capable of biased activation of the MOR, and is suitable for postoperative analgesic treatment. According to the limited information disclosed, Tegileridine is similar to compounds such as oliceridine, TRV734, and SHR9352, acting as a biased agonist preferentially activating the G protein signaling pathway rather than recruiting β-arrestin 2.

Opioid Receptors and Analgesia
Opioid receptors are membrane receptors located within the human nervous system that, upon binding with endogenous and exogenous opioid substances, can modulate pain perception and a variety of physiological functions. In the development of analgesic drugs, opioid receptor agonists play a pivotal role, primarily used for the treatment of moderate to severe, acute or chronic pain.
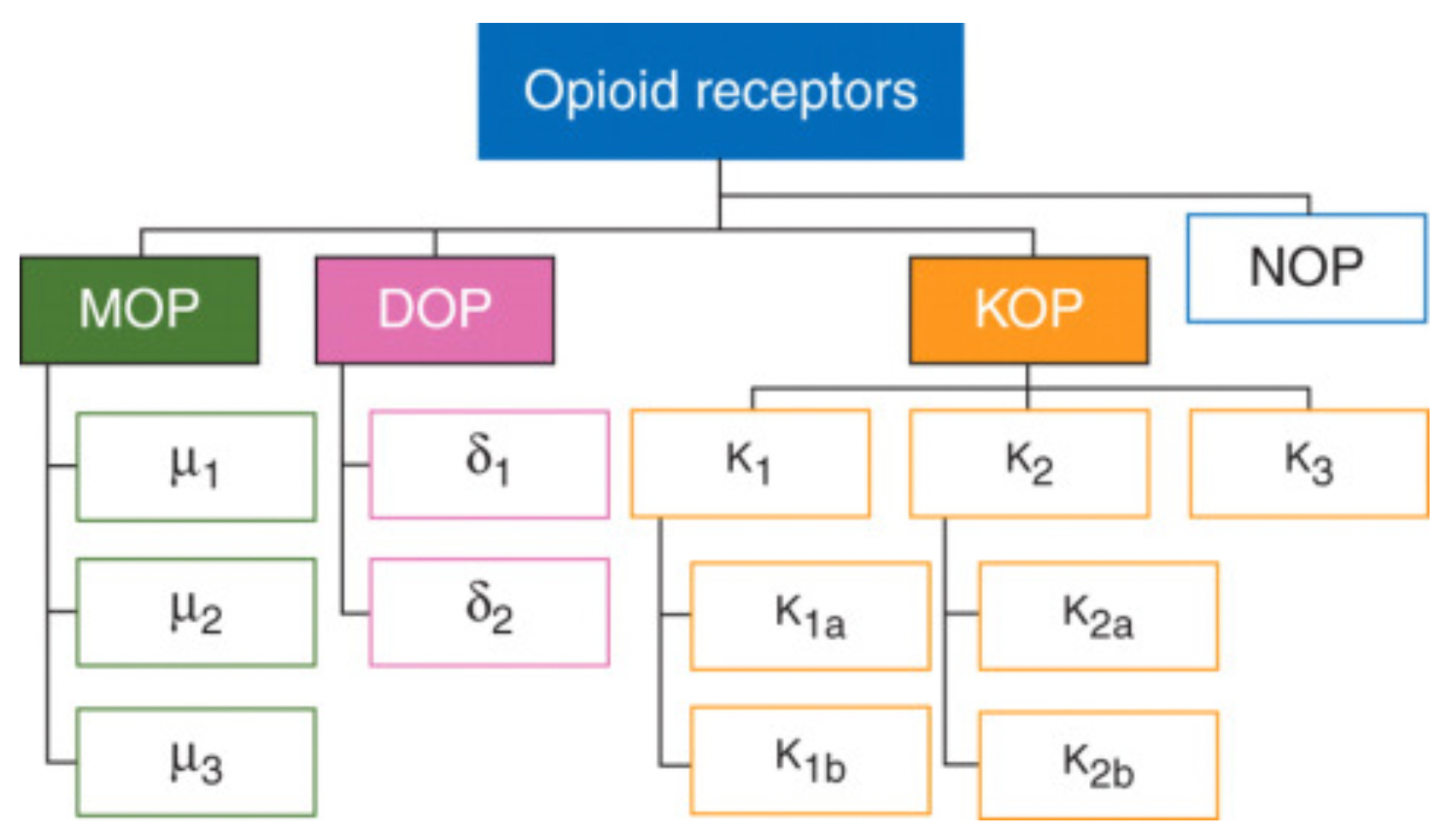
The opioid receptor family primarily consists of three members: the mu-, delta-, and kappa-opioid receptors, which belong to the G protein-coupled receptor (GPCR) superfamily and represent excellent therapeutic targets for pain control.
Mu-opioid receptor (μOR): This is the most common subtype among opioid receptors and is closely associated with various opioid drug effects, such as analgesia, sedation, respiratory depression, euphoria, and addictive potential. It is further divided into subtypes μ1, μ2, and μ3.
Kappa-opioid receptor (κOR): Exhibits high affinity for certain specific opioid drugs, such as ketocyclazocine, and has effects that differ from μOR, possibly involving analgesia, anxiety modulation, cognitive function, and potential antidepressant effects, but is also associated with potential psychotomimetic side effects.
Delta-opioid receptor (δOR): Mediates the effects of some opioid drugs, particularly with respect to pain relief, and may also be involved in emotion regulation, memory processes, and certain functions of the cardiovascular system.
Additionally, some literature mentions the NOP (nociceptin/orphanin FQ peptide receptor), which is a receptor activated by the endogenous peptide nociceptin/orphanin FQ. Although structurally similar to opioid receptors, it is usually not classified among the classical opioid receptor subtypes but is discussed as an independent member of the opioid receptor family.
Traditional opioid receptor agonists, including morphine and fentanyl, exert their potent analgesic effects mainly by activating the μ-opioid receptor. The μ-opioid receptor belongs to the G protein-coupled receptor family. When these agonists bind to and activate the G protein pathway, they inhibit the neuron's ability to transmit pain signals, thereby producing significant central analgesia.
However, the activation of opioid receptors is not limited to a single signaling pathway. In addition to the G protein pathway, opioid receptors can also activate non-classical signaling pathways through interaction with β-arrestin-2. Although β-arrestin-2 is involved in regulating receptor activity and stability, its overactivation is associated with a range of adverse reactions, such as respiratory depression, constipation, and other issues related to tolerance and dependence. This significantly limits the safety and efficacy of traditional opioid drugs in clinical applications.
Given the recognition of biased agonism theory in this field, the development of modern opioid receptor analgesics focuses on finding and designing new compounds with greater selectivity that preferentially activate the G protein pathway without causing or minimizing β-arrestin-2 mediated side effects. This is in the hope of achieving a better balance between analgesic effects and safety.
About Oliceridine
In August 2020, Trevena announced on its official website that the FDA has approved the company's New Drug Application for Oliceridine (OLINVYK, TRV130) injection for the management of acute pain in adults that is severe enough to require an intravenous opioid analgesic and for which alternative treatments are inadequate.
Oliceridine is a biased agonist of the μ-opioid receptor and is the first such biased agonist to be approved by the FDA in decades. It is expected to fulfill a significant unmet need in the field of acute pain management.
Oliceridine offers the efficacy of intravenous opioid medications with rapid onset, requiring only 2-5 minutes to take effect. Furthermore, Oliceridine does not require dose adjustment in patients with renal impairment, a large patient population that is particularly at risk of serious medical complications.
In the clinical setting, the adjustment of traditional opioid receptor agonist dosages can provide adequate analgesic effects, but may be limited by adverse reactions, including respiratory and central nervous system depression. Oliceridine's mechanism of action, through biased activation of downstream G protein signaling pathways, allows it to have lower side effects.
According to the drug safety communication issued by the FDA, the concomitant use of opioids with central nervous system depressants (including alcohol) may result in profound sedation, respiratory depression, coma, and even death.
Preclinical and Clinical Research Data on Oliceridine
The μ-opioid receptor is one of the first G Protein-Coupled Receptors (GPCRs) for which the possibility of developing biased ligands has been demonstrated, thereby improving the benefit-risk profile of existing analgesic drugs. When activated by endorphins or opioid substances, the μ-opioid receptor triggers two pathways inside the cell: the G protein pathway (primarily associated with analgesia) and the β-arrestin pathway (mainly related to respiratory and gastrointestinal effects).
In preclinical studies of Oliceridine, β-arrestin-2 knockout mice treated with morphine exhibited a stronger analgesic effect compared to wild-type mice also treated with morphine; respiratory depression was reduced by approximately 50%.
According to preclinical data, Oliceridine shows a bias towards activating the G protein signaling pathway downstream, while its agonist capability towards the β-arrestin signaling pathway is weaker.
In terms of analgesic efficacy, Oliceridine not only achieves the maximum analgesic effect comparable to the traditional labor pain medication morphine, but also demonstrates better potency than morphine in the analgesic activity studies using the rat hot plate model. Importantly, compared to morphine, Oliceridine significantly reduces respiratory depression at equianalgesic doses and has a broader therapeutic window, as highlighted in yellow.
Additionally, according to phase 2a clinical trial data released by Trevena Inc. for Oliceridine in bunionectomy surgeries, patients who received 4 mg of morphine intravenously had significantly lower pain scores (NPRS) compared to placebo within the first 3 hours post-medication. The pain scores for the groups treated with 0.5 mg and 1 mg of Oliceridine overlapped with the 4 mg morphine group throughout the entire initial dosing interval, indicating that Oliceridine exhibited approximately five times the potency of intravenous morphine upon the first administration in this study. Patients receiving the higher doses (2 mg and 3 mg) experienced a dose-related reduction in pain intensity that was also superior to placebo.
In two randomized, multicenter, double-blind, placebo-controlled phase 3 clinical trials, Oliceridine, administrated in varying concentrations, demonstrated significantly superior efficacy in treating breakthrough pain compared to placebo, and was comparable to high doses of morphine.
Tegileridine subtype selectivity and biased characteristics
According to the relevant compound patent (WO2017063509A1) submitted by Hengrui Company in 2017, compounds in the Tegileridine series show strong agonistic activity towards the Gi signaling pathway downstream of the μ-opioid receptor, while exhibiting weaker agonistic capabilities at the δ- and κ-opioid receptors, indicating that these compounds possess strong subtype selectivity.
Furthermore, this series of compounds demonstrates weaker agonistic activity for the β-arrestin signaling pathway downstream of the receptors. Overall, compounds in the Tegileridine series are highly selective and demonstrate a strong bias towards the Gi protein signaling pathway.