Cdc42 provides a new target for treating Alzheimer’s Disease
Scientists from Southern Medical University of China have discovered new evidence suggesting that the small G protein family member, Cdc42, plays an important role in the progression of Alzheimer's disease. Alzheimer's, the most common cause of dementia, is characterized by extracellular senile plaques and intracellular neurofibrillary tangles, as well as a significant decrease in the density of neuronal dendritic spines. Although Cdc42 is known to regulate synaptic plasticity and is regulated by Cdc42GAP, previously few studies have focused on its involvement in Alzheimer's disease progression.

The Role of Cdc42 in Alzheimer
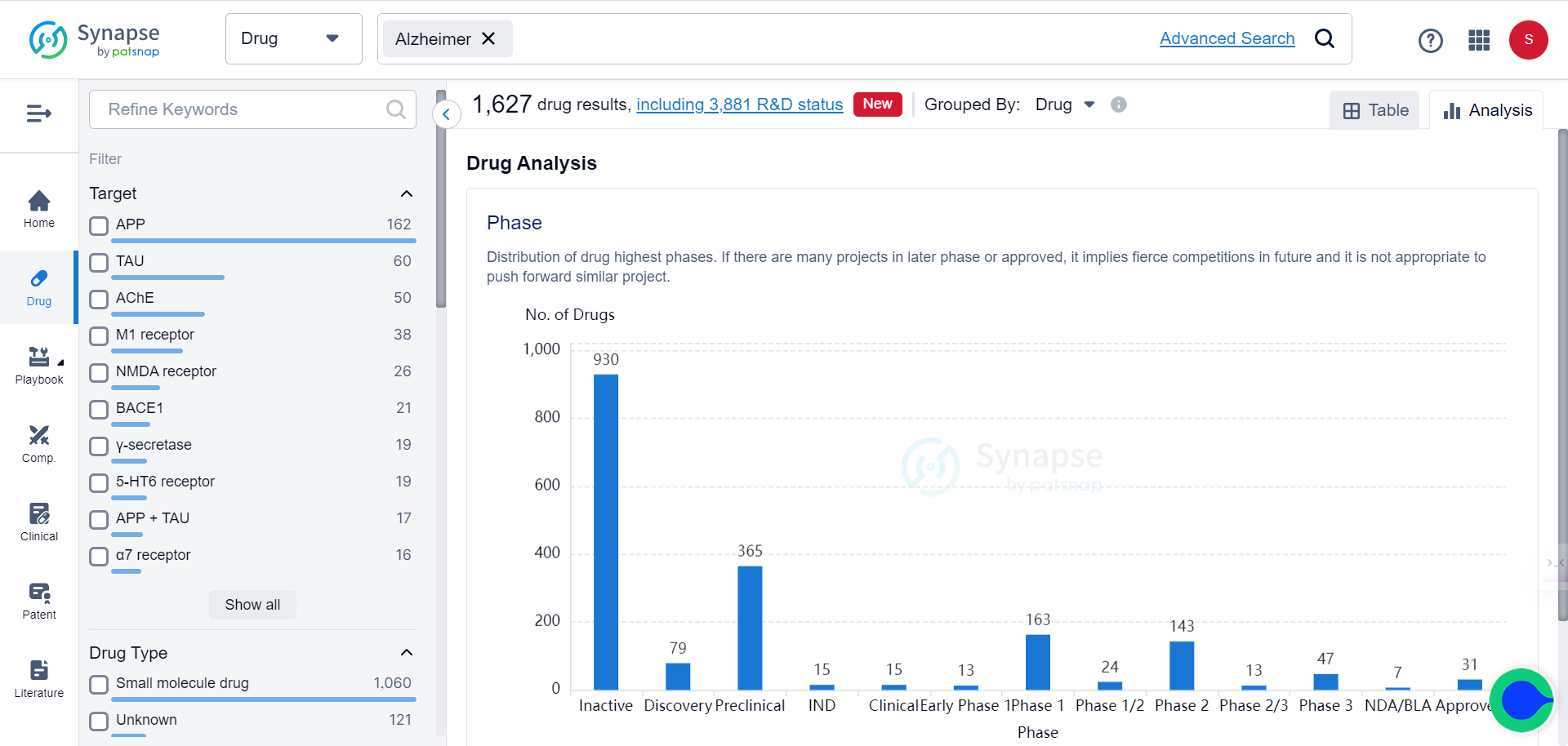
Cdc42, a signaling protein indispensable for actin cytoskeleton reorganization and cell morphogenesis, has a poorly characterized role in synaptic plasticity and behaviors like learning and memory formation. According to the Synapse database, currently there are only 2 drugs targeting Cdc24 in the market, both are Cdc24 inhibitors for treating cancers:
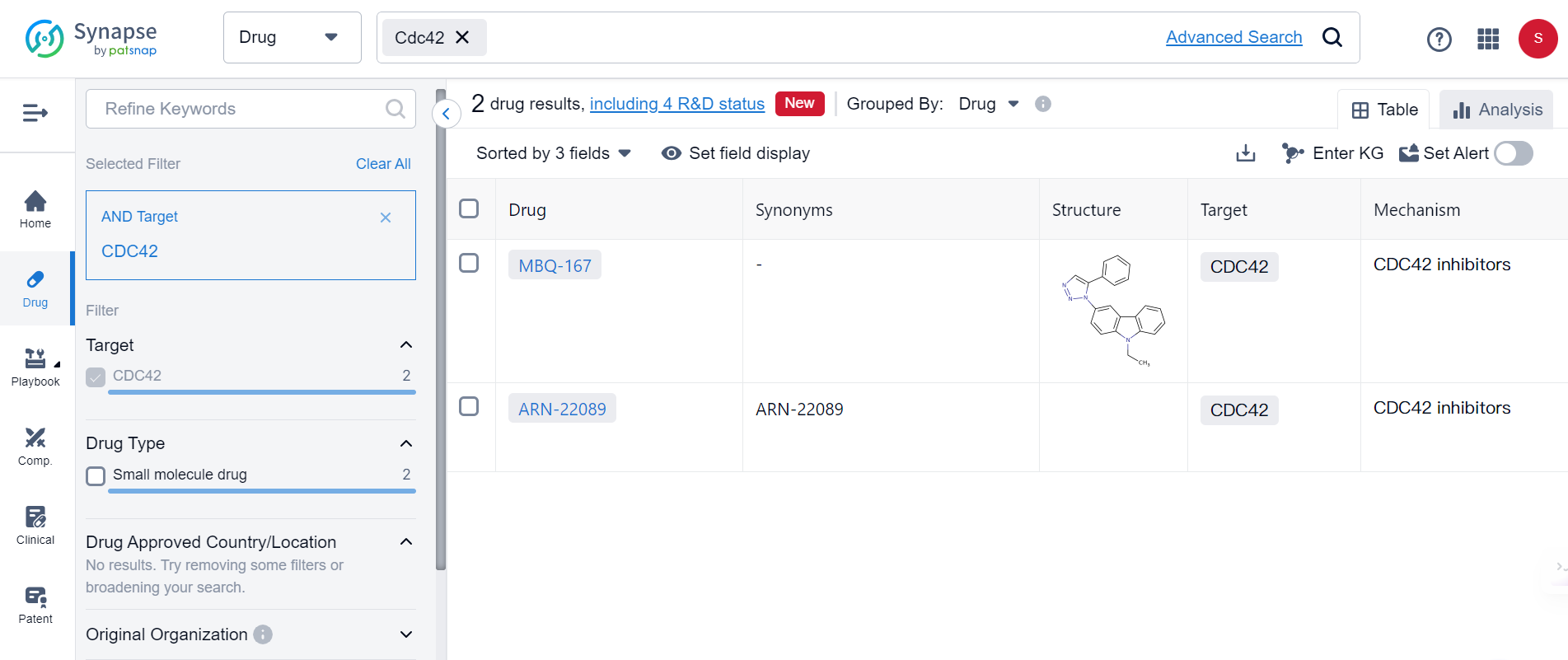
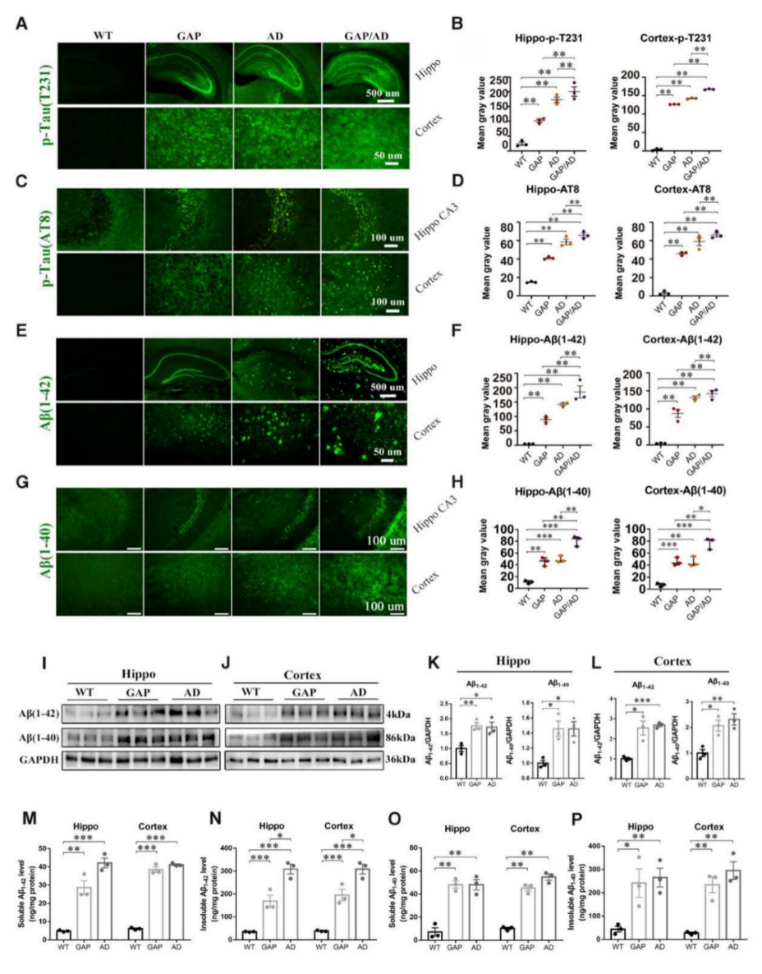
This may suggest new opportunities for developing treatments of Alzheimer. To investigate this further, scientists used a heterozygous Cdc42GAP mouse model that exhibited elevated Cdc42-GTPase activity accompanied by increased Cdc42-PAK1-cofilin signaling. They discovered that impairments in cognitive behaviors, neuron senescence, synaptic loss with depolymerization of F-actin, and the pathological phenotypes of Alzheimer's disease, including phosphorylated tau (p-T231, AT8), along with increased soluble and insoluble Aβ1–42 and Aβ1–40, were consistent with typical Alzheimer's disease mice. Interestingly, these impairments increased significantly with age.
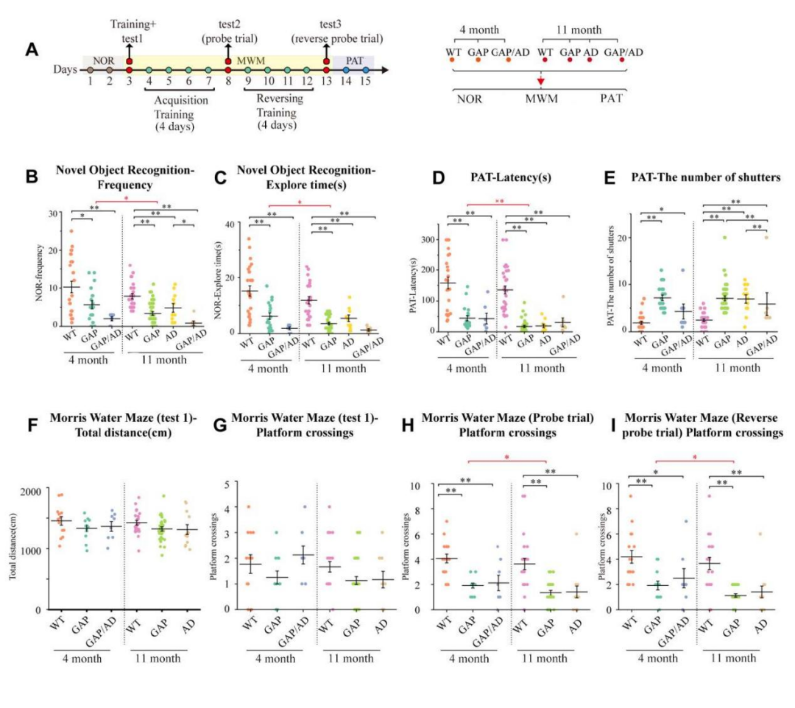
Moreover, the scientists conducted a quantitative phosphoproteomic analysis of the hippocampus of 11-month-old GAP mice. The analysis demonstrated that Cdc42GAP deficiency induces and accelerates Alzheimer's disease-like phenotypes by activating GSK-3β through dephosphorylation at Ser9 and Ser389, as well as phosphorylation at Tyr216. Furthermore, the overexpression of a dominant-negative Cdc42 in the primary hippocampal and cortical neurons of heterozygous Cdc42GAP mice was found to reverse synaptic loss and tau hyperphosphorylation.
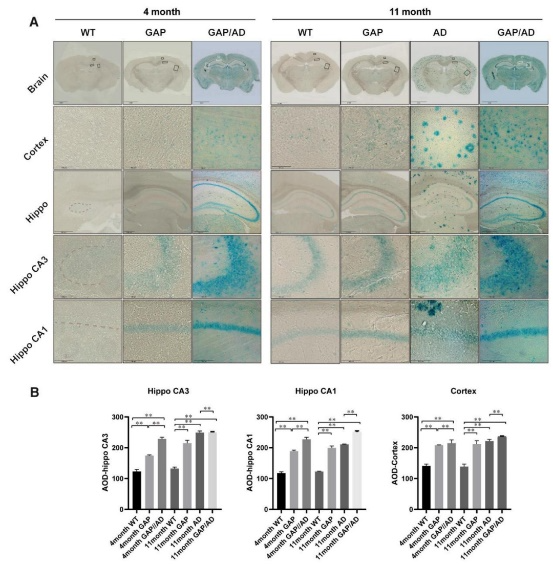
The Cdc42 signalling pathway, Aβ1–42, Aβ1–40, and GSK-3β activity were increased in the cortical sections of Alzheimer's disease patients compared to those in healthy controls. These findings suggest that Cdc42GAP is involved in regulating Alzheimer's disease-like phenotypes, such as cognitive deficits, dendritic spine loss, phosphorylated tau (p-T231, AT8), and increased soluble and insoluble Aβ1–42 and Aβ1–40, possibly through the activation of GSK-3β, and these impairments increase significantly with age.
Deficits in Remote Memory Recall
It’s not the first time Cdc42’s involvement in Alzheimer’s disease has been reported. A previous study has found that the loss of Cdc42 can lead to deficits in remote memory recall. The study used conditional knockout of Cdc42 to investigate its role in memory formation and found that postnatal forebrain deletion of Cdc42 impaired LTP in the Schaffer collateral synapses and postsynaptic structural plasticity of dendritic spines in CA1 pyramidal neurons in the hippocampus. However, the loss of Cdc42 did not appear to affect memory acquisition, but instead significantly impaired remote memory recall. The results suggest that the postnatal functions of Cdc42 may be crucial for maintaining synaptic plasticity in hippocampal neurons, which contributes to the capacity for remote memory recall. The findings could have implications for developing treatments for memory-related disorders.
The loss of memory is a common symptom of many neurological disorders, including Alzheimer's disease. The discovery that Cdc42 plays a role in memory recall could lead to the development of new therapies for these conditions.
As shown in the Synapse database, the current approach for treating Alzheimer’s disease predominantly targets APP and TAU, corresponding to two of the most prominent signatures of the condition, to varying degrees of success.
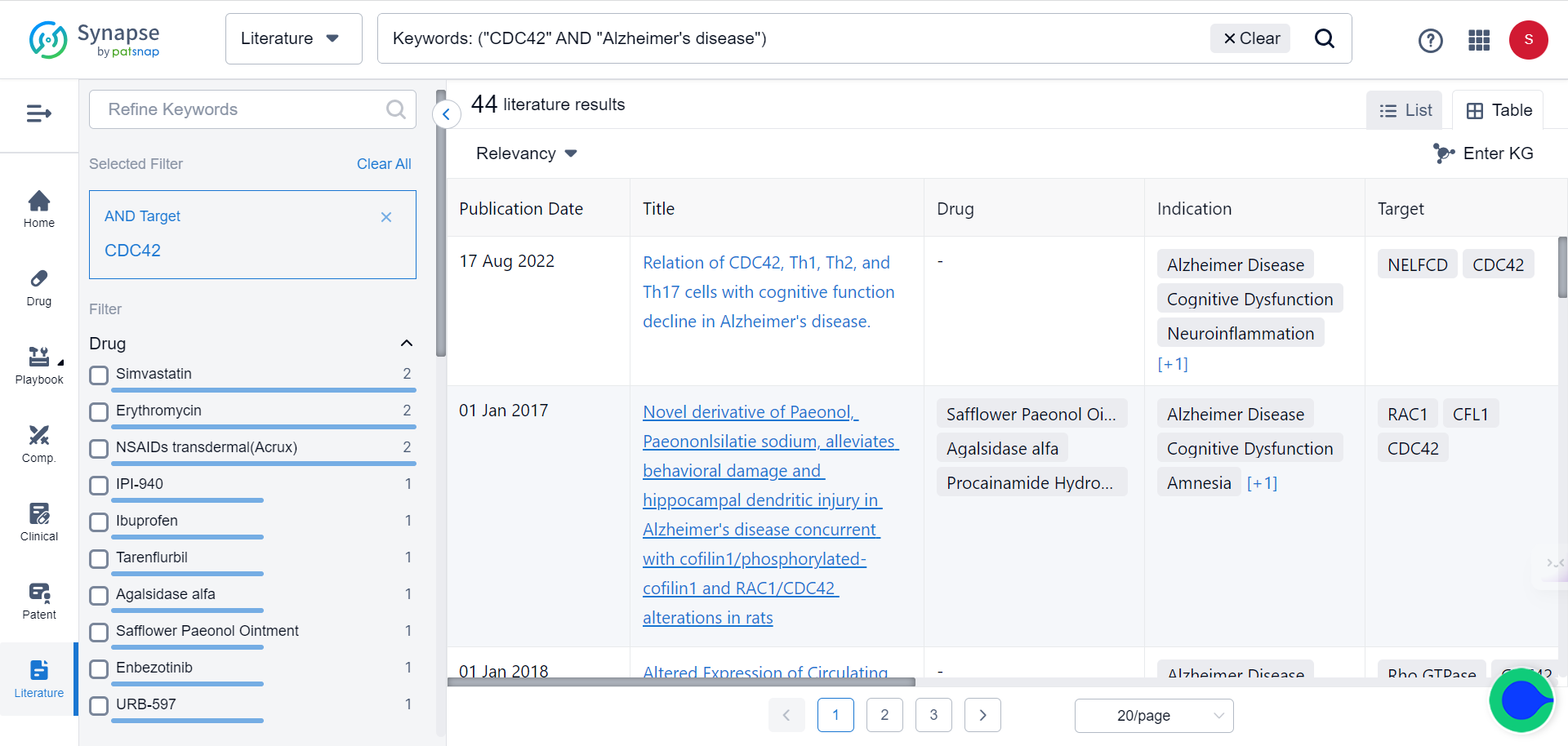
However, in recent years, Cdc42 has increasingly caught the attention of researchers finding cures for the Alzheimer’s, as demonstrated by the growing body of literature on this topic (search Synapse’s literature database with keywords CDC42 and Alzheimer’s disease):
This discovery provides new targets for Alzheimer's disease treatment and sheds light on the involvement of Cdc42 in the progression of Alzheimer's disease-like phenotypes. These findings could lead to the development of new therapies for Alzheimer's disease, which affects millions of people worldwide, and improve the quality of life for those affected by this debilitating disease.
Alzheimer and Beyond
In addition to Alzheimer's disease, researchers have identified that Cdc42 plays a role in frontotemporal lobar degeneration (FTLD), a group of neurodegenerative disorders characterized by neuronal degeneration, protein inclusions, and synaptic loss. The molecular mechanisms underlying these changes remain unclear. The research team conducted an investigation into whether Cdc42 protein levels were altered in FTLD patients and discovered that Cdc42 was increased in the frontal cortex of FTLD patients compared to age-matched controls, as well as in Alzheimer's disease patients included in the data set. However, the pool of circulating Cdc42 in the plasma was altered in FTLD patients but not in AD patients.
Interestingly, the researchers also found that the expression of Cdc42 was specifically decreased in the behavioral subgroup of FTLD patients. These findings suggest that Cdc42 may play a role in FTLD, particularly in the behavioral variant. The results offer new insights into the molecular mechanisms underlying FTLD and suggest that Cdc42 could be a potential therapeutic target.

References:
1.Zhu M, Xiao B, Xue T, et al. Cdc42GAP deficiency contributes to the Alzheimer's disease phenotype, Brain. 2023; awad184. doi:10.1093/brain/awad184
2.Saraceno, Claudia et al. Altered Expression of Circulating Cdc42 in Frontotemporal Lobar Degeneration. 1 Jan. 2018: 1477 – 1483.



