Drugging the “Undruggables”: Small Molecules for neurodegenerative disease
As the population ages, the incidence of neurodegenerative diseases such as Alzheimer's and Parkinson's are surging, presenting significant threats to public health and weighing on society. However, effective treatments for most neurodegenerative conditions remain elusive, with existing options merely providing symptomatic relief without targeting the root causes. While some biologic therapies have shown early promise in treating these diseases, developing small molecule drugs that are affordable and convenient continues to pose a major challenge.
The “undruggables”
A key reason for this difficulty lies in the "undruggable" nature of proteins involved in neurodegeneration. Unlike traditional drug targets that have enzymatic activity and binding sites for small molecules to modulate their functions, pathological proteins in neurodegenerative diseases are often misfolded, lacking sites that drugs could target, making medication development extremely hard.
Despite decades of efforts, successes in developing effective neurodegenerative disease therapies like Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS) remain scarce. The intricate disease etiology, unique target traits, and inherent obstacles in delivering therapeutics to the nervous system pose formidable challenges for drug development.
However, evidence from human genetics and animal models implicates amyloid-β (Aβ) and tau in Alzheimer's disease, α-synuclein in Parkinson's disease, TDP-43 in amyotrophic lateral sclerosis and frontotemporal dementia, and mutant huntingtin (mHTT) in Huntington's disease as central to the pathological processes and progression of these conditions. Together, they represent potential drug targets that can be addressed through various strategies.
Broadly speaking, these strategies fall into indirect or direct targeting approaches, and each category can then be further divided based on the pathways involved. Indirect approaches aim to modulate factors that influence protein levels or aggregation, while direct approaches seek to target pathogenic proteins or their interactions directly.
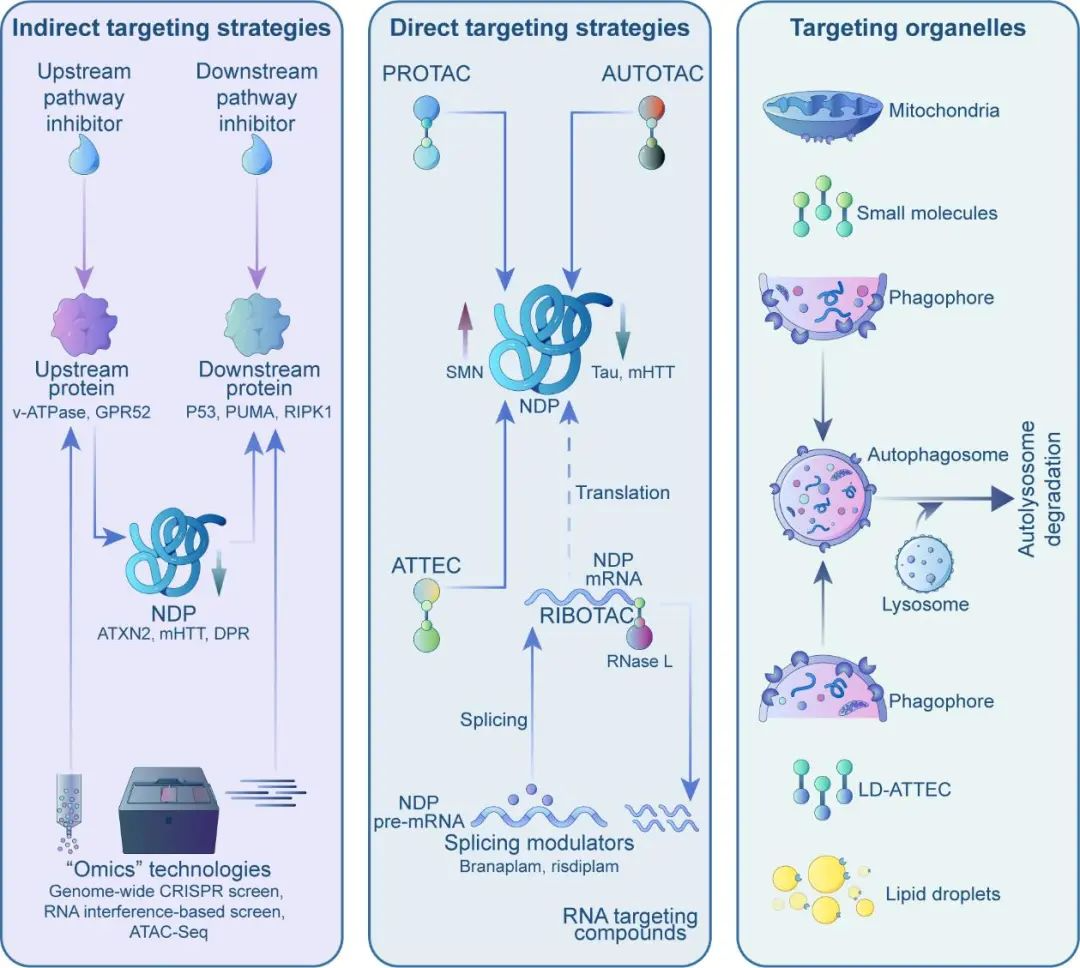
Indirect Targeting Strategies
While directly targeting "misfolded" pathogenic proteins is difficult, modifying upstream or downstream factors may be more feasible. For instance, through omics screens, researchers identified the upstream genes v-ATPase and GPR52 that modulate levels of Ataxin 2 (implicated in ALS and SCA2) and mutant huntingtin (mHTT). Follow-up studies have demonstrated that the utilization of small molecules to inhibit v-ATPase and GPR52 can efficiently and securely decrease Ataxin 2 and mHTT expression in both mouse and human neurons, leading to a notable enhancement of disease phenotypes associated with the condition.
This approach involves designing drugs that specifically target the more "druggable" upstream modifiers of pathogenic proteins. However, as these upstream genes serve normal functions, the inhibition or activation of them can potentially lead to side effects, thereby limiting the effectiveness of this strategy. It will be crucial to establish a therapeutic window that enables the reduction of disease proteins without interfering with the essential functions of the modifiers.
Alternatively, targeting downstream molecular pathways can also offer opportunities. Misfolded proteins can elicit neurodegeneration through complex mechanisms. Researchers have discovered that the inhibition of the p53-Puma pathway, which is activated in both ALS and FTD, effectively prevented neuronal death in both cell and animal models. These findings suggest that Puma inhibitors may have therapeutic potential for the treatment of these disorders.
Likewise, the inhibition ofRIPK1, a crucial mediator of cell death and inflammation pathways in neurodegenerative disorders, has demonstrated safety both in preclinical and clinical studies. This presents opportunities for the treatment of diseases such as Alzheimer's, ALS, and multiple sclerosis. Overall, these strategies aim to target the aberrantly activated pathways in neurodegeneration, potentially with fewer side effects.
However, targeting individual downstream proteins may also have limitations, since misfolded proteins trigger related diseases through diverse signaling routes, potentially interfering with or compensating for drugs against these pathways.
Direct Targeting Strategies
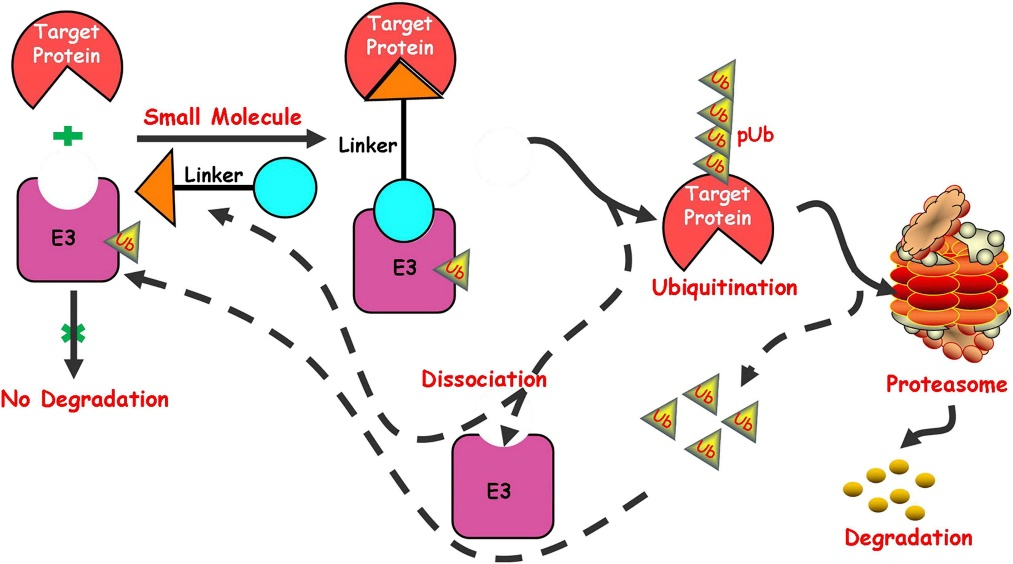
Although directly reducing misfolded protein levels with small molecules is unfeasible, targeted protein degradation technologies, such as PROTACs and AUTOTACs, provide viable alternatives. These technologies function similarly, comprising three components: a ligand that binds to the target protein, a ligand for cellular degradation machinery components, and a linker that connects them.
By acting as "molecular bridges," they facilitate interactions between targets and degradation elements, leading to the breakdown of the target. However, PROTACs and AUTOTACs are large, chimeric molecules with suboptimal drug properties. As chimeras, they often exhibit poor blood-brain barrier penetration, which poses challenges for treating neurodegenerative diseases.
Recently, researchers proposed a new autophagy-targeting "molecular glue" strategy called ATTEC (AuTophagy-TEthering Compounds), which effectively addresses these issues. Studies have demonstrated that these "molecular glues" can degrade mutant huntingtin without affecting normal huntingtin, thereby rescuing Huntington's disease phenotypes in cells, flies, and mice. With molecular weights ranging from 250-500 Da, some "molecular glues" can cross the blood-brain barrier, exhibiting enhanced drug-like properties and representing potent tools for neurodegenerative drug discovery.
Furthermore, "molecular glues" have uses beyond protein degradation; they can also elevate protein levels. For disorders caused by haploinsufficiency, upregulating wild-type protein levels may be desirable. In such cases, "molecular glues" can bind wild-type proteins to prevent their degradation by deubiquitinases.
Unlike directly targeting neurodegenerative proteins, another strategy uses small molecule drugs against their encoding RNAs. For example, small molecules can alter mRNA splicing. Novartis reported an orally available small molecule, branaplam, that lowers mutant huntingtin levels by introducing a termination codon into mRNA via splicing modulation, decreasing mutant and normal HTT production. This compound was previously reported to increased SMN protein in spinal muscular atrophy (SMA) by stabilizing SMN2 gene products, and a similar drug from Roche was approved by FDA in 2021 for SMA.
In addition to the aforementioned strategies, Matthew Disney's team has developed RNase recruitment bivalent oligonucleotides (RIBOTACs) that offer new RNA degradation options. RIBOTACs interact with target RNAs to recruit RNase L for degradation, and preclinical studies have shown their effectiveness in degrading RNAs with expanded repeats, thereby alleviating ALS/FTD phenotypes. However, the recognition of specific RNAs by small molecules is limited to specialized structures, usually in GC-rich repetitive sequences, and RIBOTACs are relatively large and unable to penetrate the brain effectively. Nonetheless, these results pave the way for future research directions.
Aberrant organelles in neurodegeneration may also provide insights into drug development. Mitochondrial damage and lipid accumulation are prominent features, and while organelles have been difficult targets in the past, recent advancements have expanded degradation technologies to include organelles. Methods to degrade mitochondria (AUTAC4) and lipid droplets (LD·ATTEC) have been developed, but further studies are necessary to determine if these compounds can ameliorate disease phenotypes.
Although developing small molecule drugs for neurodegenerative diseases is crucial, it has been challenging to target "undruggable" proteins. However, recent technological advancements have provided potential strategies for engaging with these difficult targets, offering hope for patients. Should these strategies prove successful, they could lead to transformative breakthroughs in neurodegenerative drug discovery and inform the targeting of "undruggable" proteins in other diseases as well.

Reference
Junmei Lu, Zhaoyang Li, Aaron D. Gitler, Boxun Lu. Drugging “undruggable” neurodegenerative disease targets with small molecules. Science Bulletin 2023.



