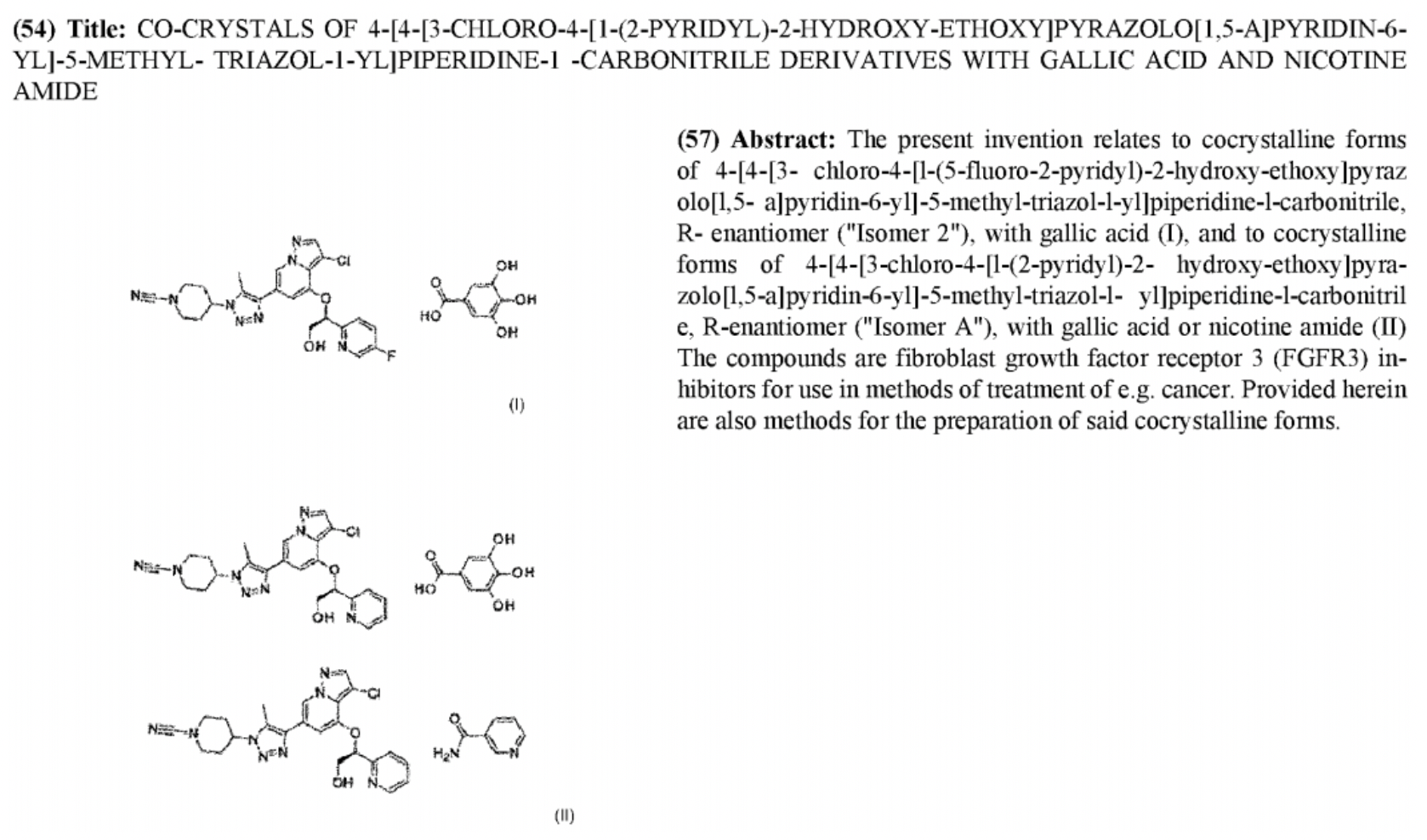
Eli Lilly Reveals the Crystal Form Patent of FGFR3, Unveiling the Structure of LOXO-435
The pharmaceutical giant Lilly, valued at $700 billion, publicly disclosed a WO patent on March 14, 2024. This patent is for a crystalline salt form of an FGFR3 selective inhibitor, as shown in the figure below. It covers the crystalline salt form of a class of pyrimido[4,5-b]pyrazine small molecule compounds. Based on Lilly's strategic development around the FGFR3 target, one of the two compounds illustrated is very likely the structure of LOXO-435. The main difference between the structures of these two compounds is the presence or absence of an F substitution at the meta position of the pyridine segment.
In April 2019, Erdafitinib was approved, becoming the first FGFR selective compound approved by the FDA for second-line treatment of metastatic urothelial carcinoma. It is an effective orally administered pan-FGFR inhibitor, with IC50 values in the low nanomolar range for all members of the FGFR family (FGFR1 to FGFR4). Pemigatinib, developed by Incyte Pharmaceuticals and approved in 2020, is an oral, potent FGFR1-3 selective inhibitor and the first FDA-approved targeted therapy for cholangiocarcinoma. Infigratinib, developed by QED Therapeutic and indicated in clinical trials primarily for chemotherapy-refractory FGFR2 fusion-positive cholangiocarcinoma, is an oral, ATP-competitive, selective FGFR1-3 inhibitor. However, common treatment-related adverse events (TRAEs) for the three approved FGFR inhibitors mentioned above, as seen in clinical trials, include hyperphosphatemia, dry mouth, fatigue, skin changes, nail changes, and ocular disease.
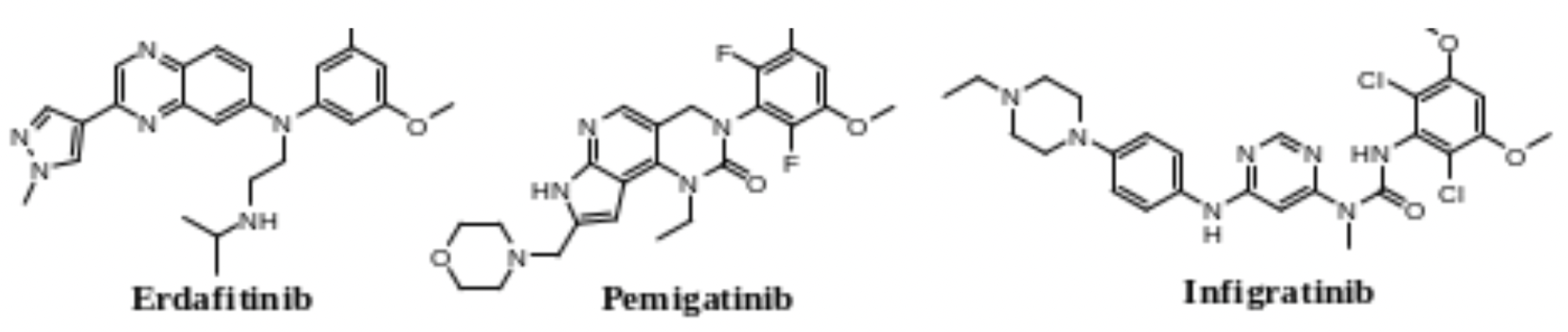
The development of pan-FGFR inhibitors as well as FGFR1-3 specific inhibitors has been successful; however, off-target side effects have informed the direction of subsequent development. For instance, FGFR1-mediated hyperphosphatemia is a dose-limiting toxicity of pan-FGFR inhibitors; cutaneous/ungual, ocular, and perioral toxicities mediated by FGFR2 lead to chronic intolerance of pan-FGFR inhibitors; domestic and international companies are dedicated to developing highly selective inhibitors targeting precise subtypes of FGFR2, FGFR3, FGFR4 and second-generation FGFR inhibitors that overcome resistance to existing FGFR inhibitors.

According to the description in patent WO2024054814A1, the crystalline salt form molecule protected by Eli Lilly has been previously disclosed in patents WO/2022/187443 and US2023/0095122A1, which have published the synthesis methods. Examples number 45 and 46 in WO/2022/187443 correspond to two isomers with pyridine carrying a fluoro substituent; example number 151 is the racemate of pyridine without a fluoro substituent. The patent only provides a general range for activity, indicating that selectivity is over 10-fold.
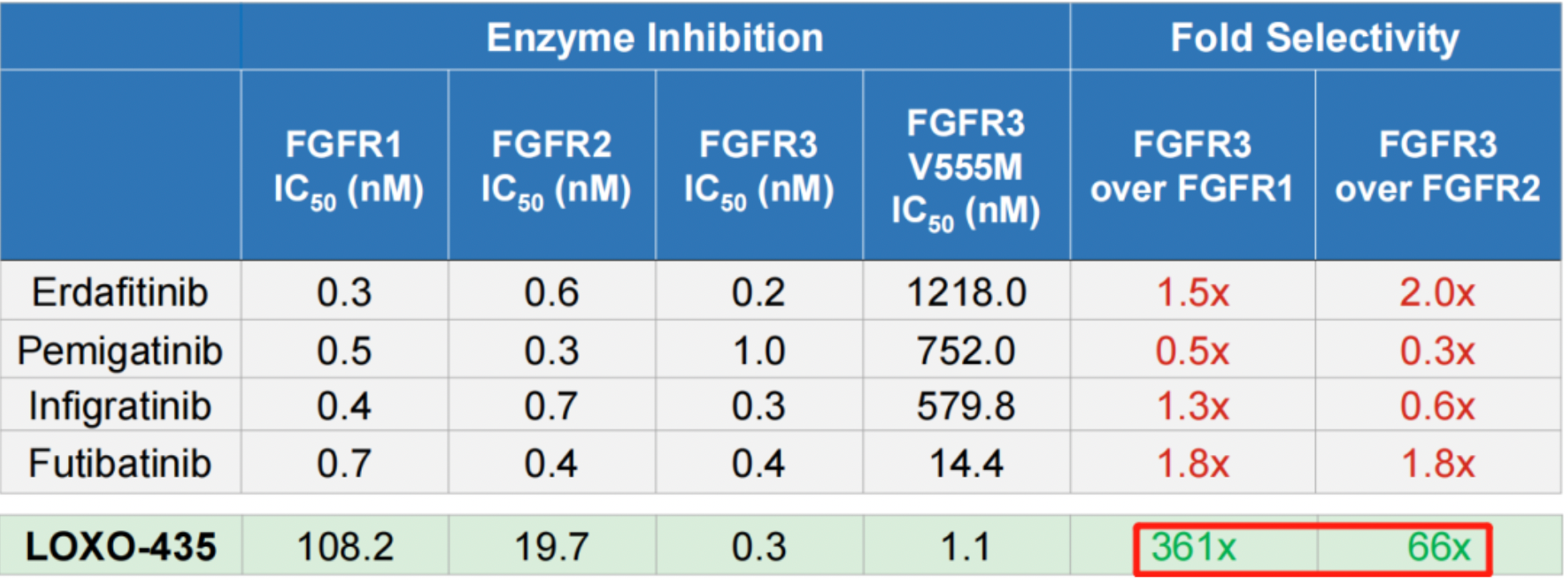
Although the selectivity of the compound in the patent is not specified in exact multiples, more biological data are disclosed in the poster. At the 2021 AACR conference, Eli Lilly released preclinical data for LOXO-435, and the Figure below demonstrates that LOXO-435 has strong efficacy and high selectivity against FGFR3 and FGFR3 V555M kinase, while retaining certain activity against FGFR1 and FGFR2. The selectivity for FGFR1 and FGFR2 isoforms reached 361-fold and 66-fold, respectively. Cellular activity data shows that LOXO-435 has an IC50 of 15.1 nM against the FGFR3-TACC3 (RT112) mutant. The selectivity for UMUC-14 over DMS114 reached 374-fold.
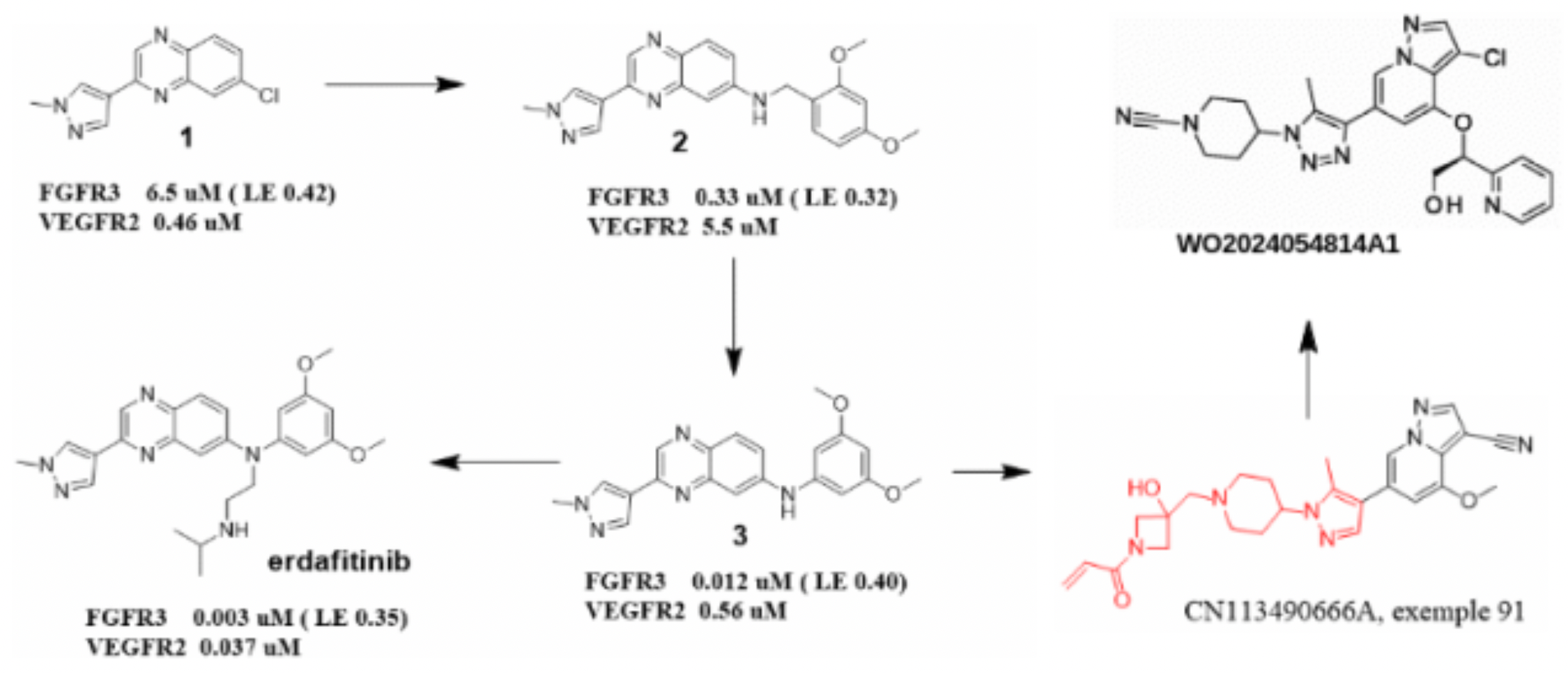
The design philosophy for the aforementioned FGFR3 selective inhibitor is speculated as follows: Based on the discovery of erdafitinib, initially, a compound with a pyrazole moiety integrated into a quinoline scaffold, Compound 1, was identified from a compound library of VEGFR2 inhibitors. This compound exhibited good selectivity towards FGFR3. By attaching an amino-substituted meta-dimethoxyphenyl ring to the right side of the benzene ring fragment in Compound 1, Compound 2 was obtained. The dimethoxyphenyl ring of Compound 2 occupies the hydrophobic pocket within the ATP-binding site through van der Waals interactions, with one of the methoxy groups forming a hydrogen bond with Asp641. Subsequent optimization of the flexible amino chain led to the development of Erdafitinib. Extending and further optimizing the pyrazole moiety resulted in the red key pharmacophore fragment shown in Figure 5, which ultimately led to the discovery of an FGFR3 selective inhibitor, specifically Compound 91 in the patent CN113490666A. Enzymatic activity assays demonstrated that FGFR3 had selectivity over FGFR1 and FGFR2 by factors of 142 and 9.5, respectively. In cellular activity assays, FGFR3's selectivity over FGFR1 was 22.7-fold. Retaining the pyrazole piperidine linker in the red fragment, introducing a cyano substitution, and derivatizing the methoxy substituent in the parent nucleus resulted in the potential PCC molecule, and substitutions at this position are also likely to enhance selectivity towards other FGFR subtypes.
Searching for Eli Lilly's clinical molecule LOXO-435 in the Synapse database yields the following information. This is a public clinical disclosure. The primary objective is to learn more about the safety, side effects, and efficacy of LOXO-435. LOXO-435 can be used for the treatment of cancers of the urinary system cell lineage and other solid tumors with specific genetic alterations, known as FGFR3 gene mutations. This is an open-label, multi-center, Phase 1a/b study, and involves participants with advanced solid tumors that exhibit FGFR3 alterations, including metastatic urothelial carcinoma (UC).
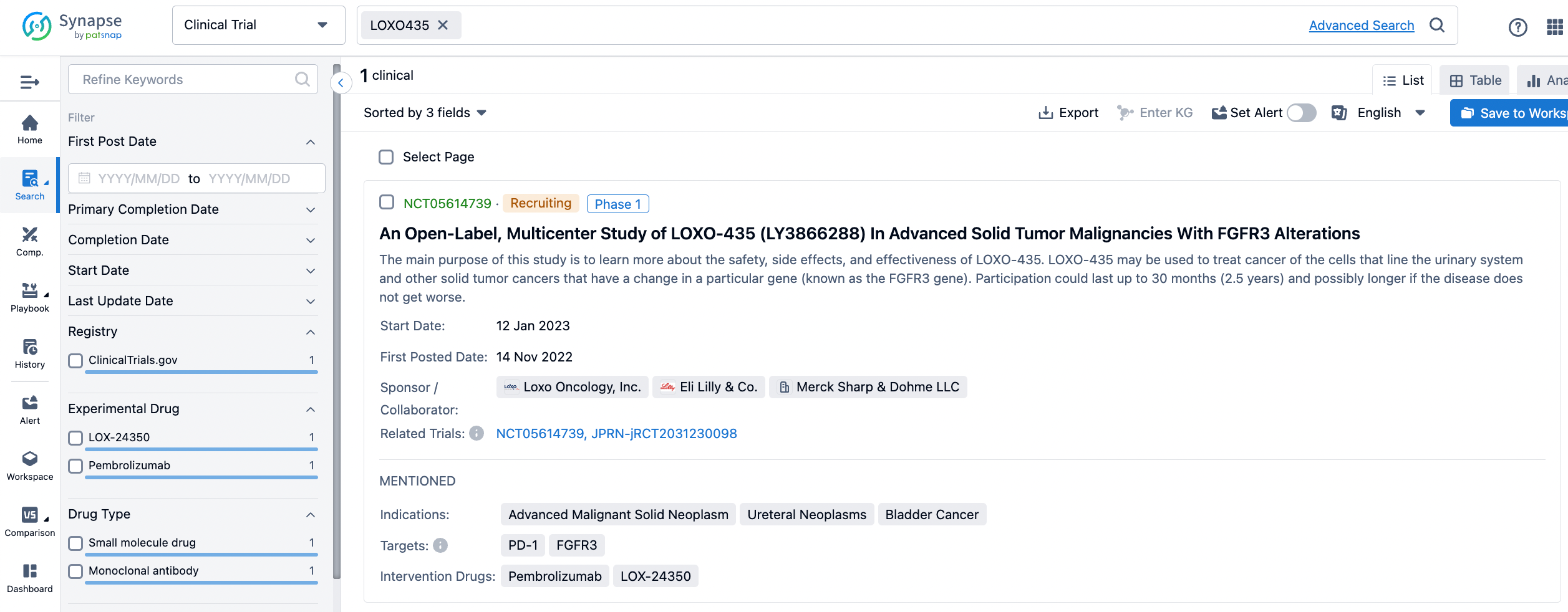
The study will be conducted in two phases: dose escalation and dose optimization (1a), and dose expansion (1b). Phase 1a will include up to two cohorts to evaluate the safety, tolerability, and pharmacokinetics of LOXO-435, in order to determine the recommended Phase 2 dose (RP2D) (or the optimal dose). Phase 1b will consist of four dose-expansion cohorts with participants pre-specified for FGFR3 alterations to evaluate the efficacy and safety of LOXO-435 at the RP2D. One of the cohorts will involve the combination treatment of LOXO-435 with Merck's Keytruda monoclonal antibody.
FGFRs, comprised of four receptor subtypes (FGFR1 to FGFR4), consist of three distinct structural domains: the extracellular ligand-binding domain, single-pass transmembrane domain, and intracellular tyrosine kinase (TK) domain. Upon binding with fibroblast growth factor (FGF) ligands, the activated FGFR phosphorylates several downstream signaling proteins including PI3K-AKT, RAS-MAPK, and STAT. Therefore, FGFR plays a crucial role in numerous intracellular processes such as development, differentiation, survival, migration, and angiogenesis. However, dysregulation of FGFR can lead to constitutive activation, which is closely associated with the development of several cancers, including FGFR2 alterations in 10 - 16% of intrahepatic cholangiocarcinomas and FGFR3 alterations in 20% of advanced urothelial carcinomas, among others, with additional abnormal subtypes depicted in the classification below the text.
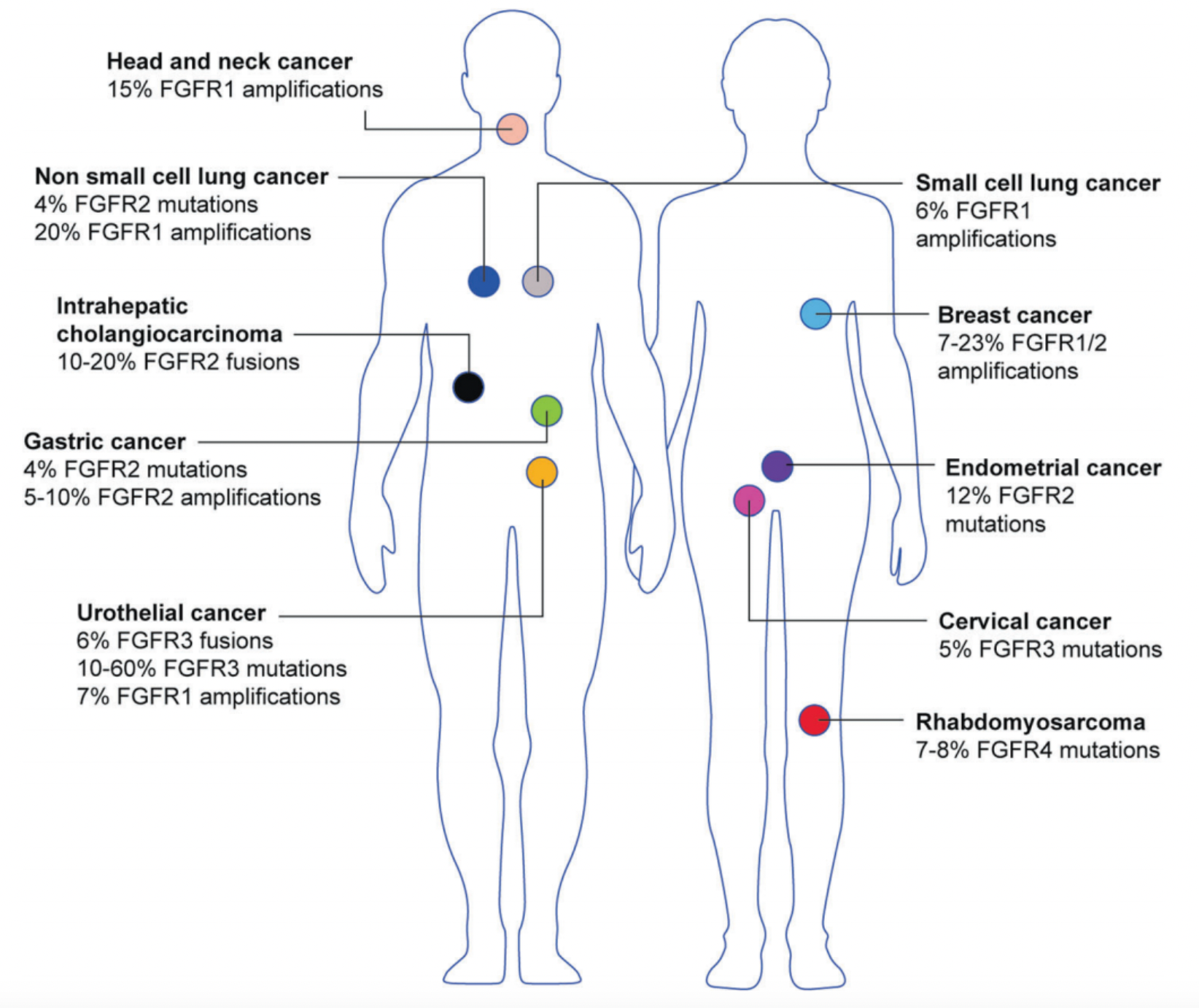
We await the disclosure of clinical data for LOXO-435 by Eli Lilly, a selective inhibitor of FGFR3 that potentially avoids the dose-limiting toxicities—such as hyperphosphatemia and other chronic intolerances—associated with inhibiting FGFR1 and FGFR2. There is a significant unmet clinical need for treatments that can offer patients safer therapeutic options. It is hoped that Eli Lilly's potent and selective inhibitor will soon be available on the market, bringing a blessing to those afflicted.

Reference:
Loxo Oncology;Preclinical characterization of LOXO-435 (LOX-24350), a potent and highly isoform-selective FGFR3 inhibitor; AACR-NCI-EORTC VIRTUAL INTERNATIONAL CONFERENCE Date: October 7, 2021.



