Exploring the Chemical Diversity of Macrocyclic Oligoamides
On April 25th, David Baker's research team from the Institute for Protein Design at the University of Washington published an article in the journal "Science" titled "Expansive discovery of chemically diverse structured macrocyclic oligoamide". This paper reported an innovative computational approach that successfully achieved the design and prediction of macrocyclic oligoamides composed of alpha, beta, gamma, and other non-natural amino acid backbone chemical structures.

In this study, using the method developed by the authors, they successfully predicted 14.9 million closed cyclic structures composed of over 42,000 different monomer combinations. The researchers chemically synthesized 18 macrocyclic compounds predicted to form a single low-energy state and determined their three-dimensional structures via X-ray crystallography or nuclear magnetic resonance techniques. Out of these 18 compounds, 15 had structures very close to the designed models. To demonstrate the therapeutic potential of these macrocycle designs, the researchers also developed specific inhibitors targeting three popular protein targets. By exploring a vast space of conveniently synthesizable, drug-like macrocyclic compounds, this research significantly enhances structure-based drug design.
In nature, there exist macrocyclic compounds constructed from atypical amino acids such as alpha, beta, gamma, and delta, along with various types of heterocyclic compounds. These compounds exhibit a broad range of biological activities, spanning antifungal and antibiotic properties to anticancer cytotoxicity and analgesic effects. Although these macrocyclic molecules demonstrate diverse biological activities in nature, the exploration of their structural space in the drug development process by scientists is far from complete, largely limited to known structural variants found in nature. The base structures and design concepts of most synthetic macrocyclic drugs approved for clinical treatment often originate from natural core structures. This implies that there is a vast amount of unexplored chemical space awaiting discovery through innovative methods and technologies, to design more macrocyclic compounds with novel activities for combating various diseases.
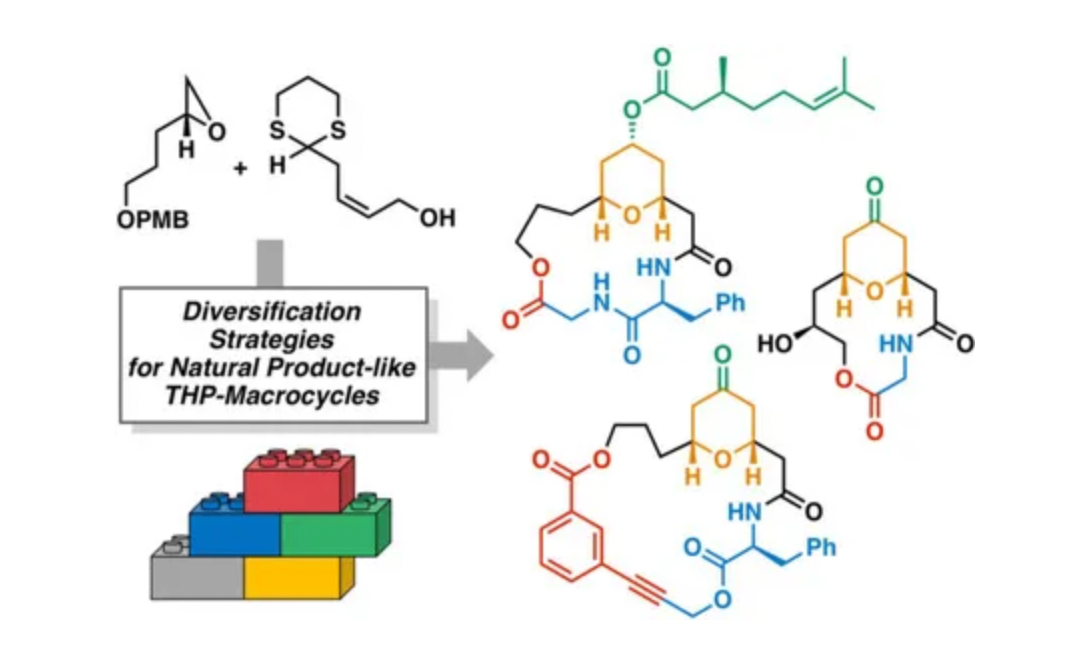
In recent years, the rapid development of computer hardware and software technology has enabled the rapid and comprehensive exploration of potential macrocyclic compound space, greatly advancing the discovery of compounds with unique biological activities similar to natural products. However, current methods are limited by the diversity of chemical structures utilized. Enumerative methods that rely on random sampling of main-chain torsion angles to identify macrocycle-closing conformations become computationally challenging when trying to construct macrocycles composed of more than one main chain building block. Taking just the 22 main-chain building blocks mentioned in the text as an example, nearly 60,000 different combinations of two, three, and four residues can be formed. Each of these combinations can be further diversified by millions of different side-chain combinations. Thus, the chemical space is so immense that it is impracticable to use explicit conformational sampling techniques directly to identify those linear sequences that can close into cyclic structures.
In this study, the researchers utilized a vast collection of 130 monomers and their corresponding stereoisomers. The team developed a computational strategy capable of efficiently exploring the potential of tri- and tetra-macrocyclic structures constructed from a variety of non-conventional amino acids and other chemical groups. Initially, the researchers innovated the computational and storage methods for each monomer conformation. They computed the low-energy conformations of each monomer and determined their optimal forms on the potential energy surface. All rigid transformations of these monomers and their dimers were computed and stored in a hash table. Using the hash table, the system systematically searches for two entries whose corresponding rigid transformations combine nearly to zero, indicating that these two segments can form a closed-loop structure. This transformation-based method avoids the direct construction and evaluation of closure for all possible backbone coordinates, significantly reducing the computational complexity.

The research team used the aforementioned method to systematically search for closed macrocyclic compounds composed of 3 to 4 residues from a vast collection containing 130 types of monomers and their corresponding stereoisomers. These monomers were categorized into 22 different chemical types (chemotypes), each distinguished based on the number of main chain atoms and their hybridization states. A hash table of nearly 9 billion low-energy conformations was constructed, and the combinations of all monomer-dimer, dimer-monomer, and dimer-dimer were systematically traversed to generate a collection of macrocyclic compounds that met the closure conditions. Compared to the approach of directly enumerating all possible combinations to check for closure, which would require constructing about 10^19 macrocyclic compounds, using the hash method reduced the complexity of evaluating these combinations from O(n^2) to O(n), significantly enhancing the speed and feasibility of the search. Ultimately, the researchers used this methodology to predict approximately 14.9 million closed macrocyclic compounds composed of various combinations of chemical type residues ranging from 3 to 32 members. Further, they experimentally validated the structure of some of these compounds, proving the effectiveness of the method. This aids in the discovery of novel macrocyclic compounds with potential pharmacological activity, thereby advancing structure-based drug design research.
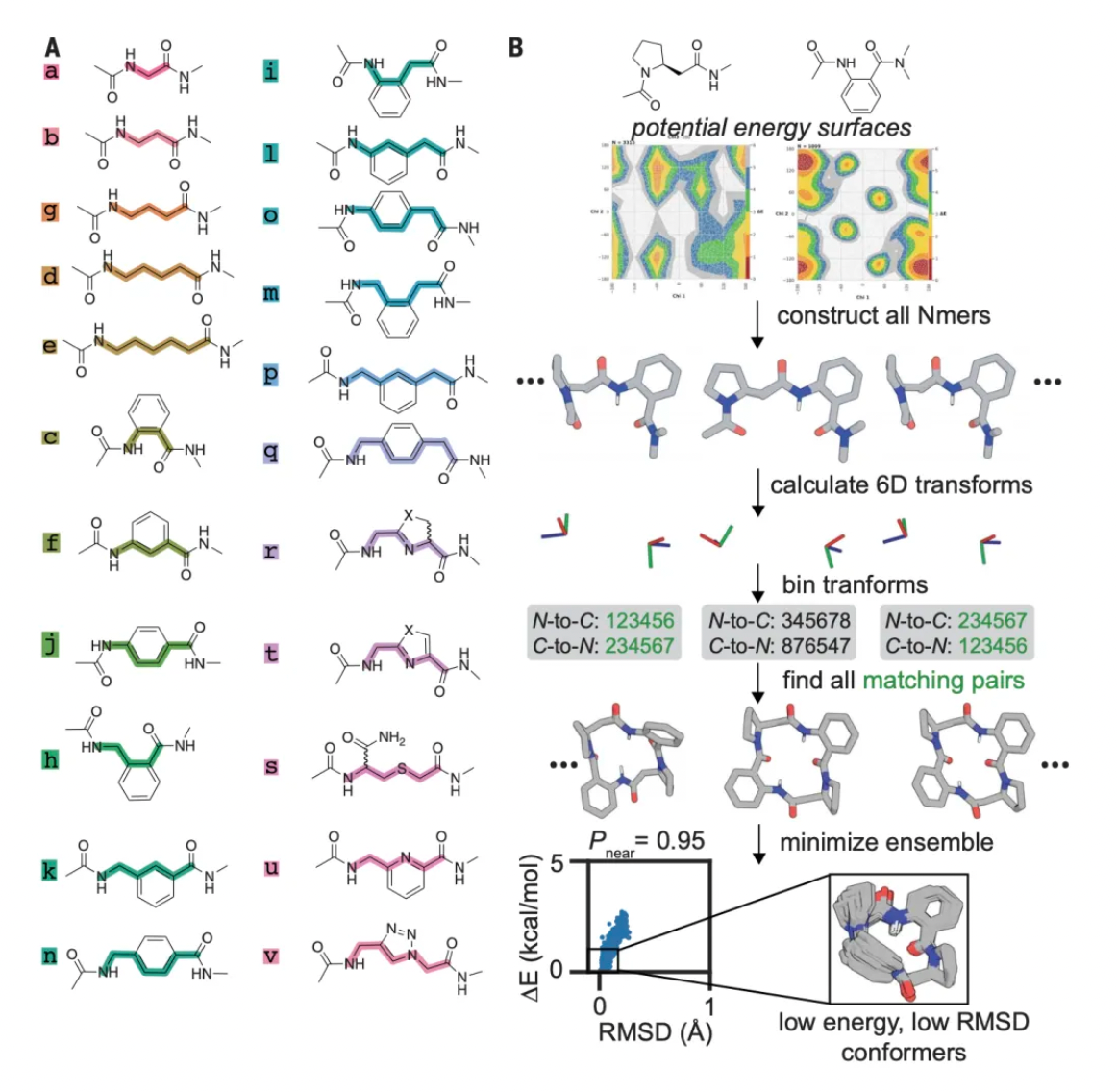
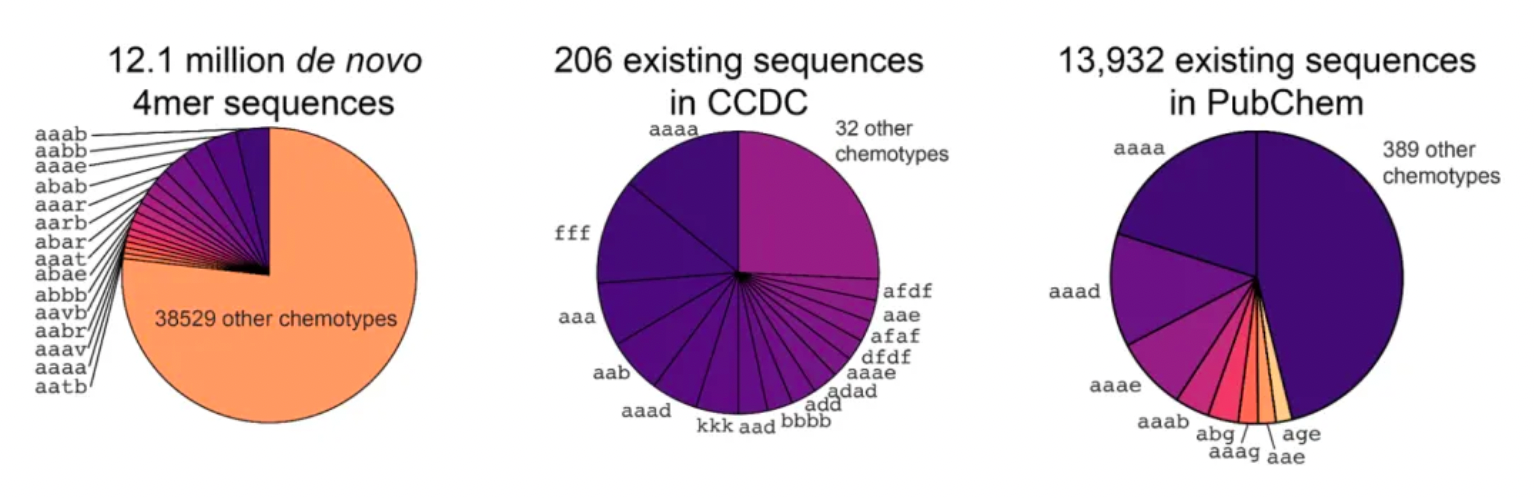
After systematically analyzing hash tables, researchers discovered 14.9 million closed macrocyclic compounds containing rings of 9 to 32 members, which belong to 3,494 trimeric and 38,544 tetrameric chemotypes. The chemical space covered by the trimeric and tetrameric chemotypes has been greatly expanded compared to previous studies. Specifically, the data shows that there are 206 compounds from 23 chemotypes with high-resolution structural data in the Cambridge Structural Database (CSD); in the PubChem database, however, 13,932 compounds belonging to 397 different chemotypes are recorded.
For each chemotype, the research team adopted a method to quantify the three-dimensional structural diversity. The researchers categorized the torsion angles of the macrocycle main chains into 60-degree angle intervals (bins) and represented each macrocycle conformation as a string of these angle intervals.
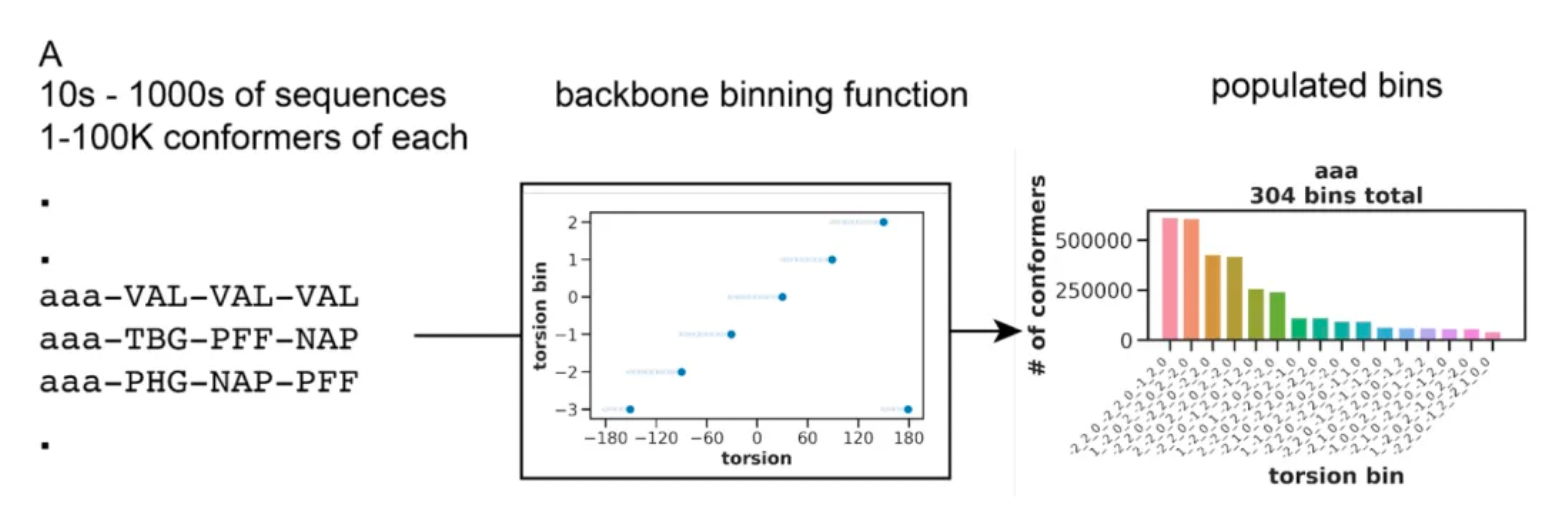
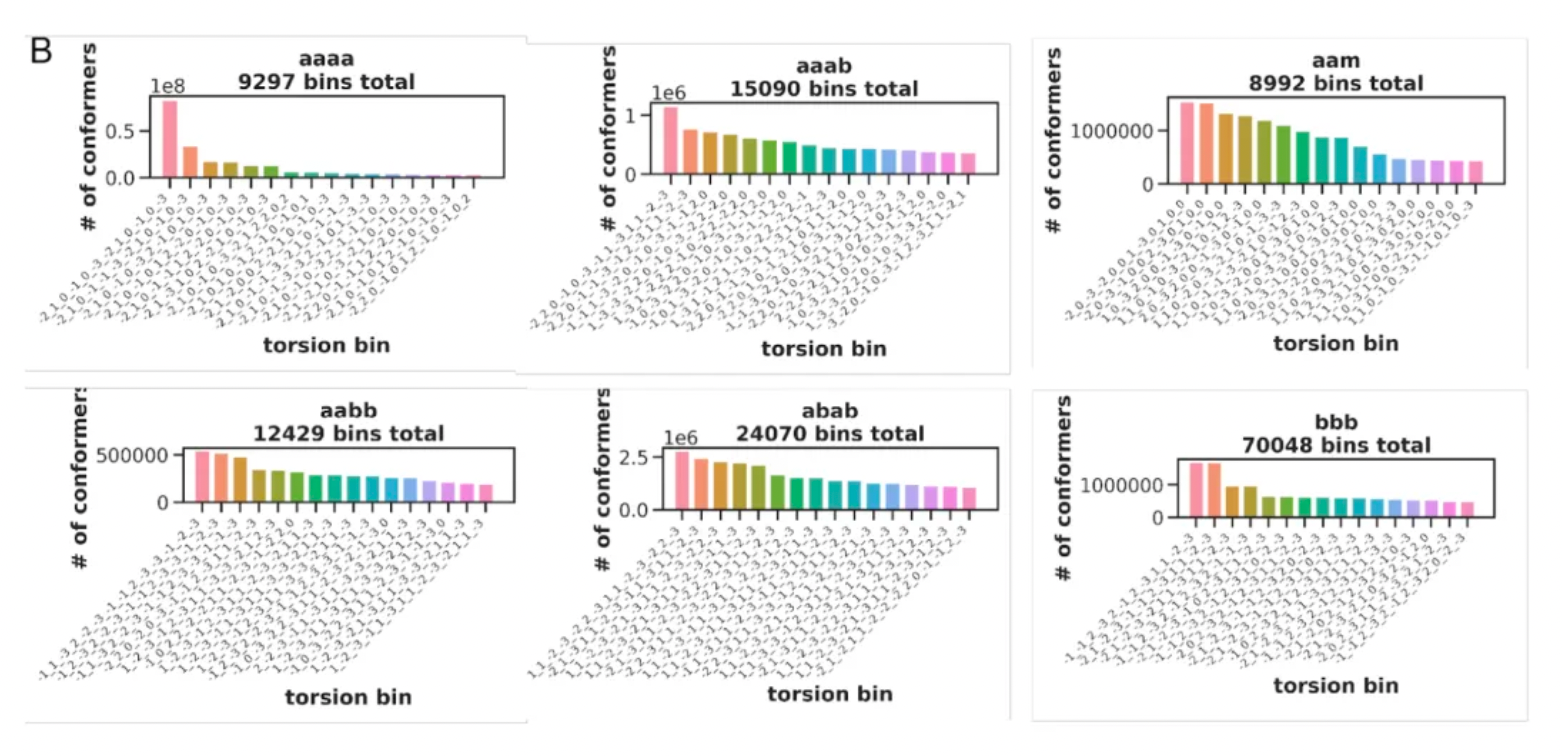
For every macrocyclic molecule belonging to each chemical type, although thousands of different conformations may be generated, in most cases, these conformations are concentrated in only a few groups of angular interval strings. This likely reflects a preference in the torsional orientations of the amino acid residues that make up the macrocycle.
By conducting a Principal Moment of Inertia (PMI) analysis on the macrocyclic conformations under all sample angle intervals for each sequence, the results indicate that different macrocyclic chemical types tend to occupy different regions in shape space. In certain special cases, such as the macrocycles of specific chemical types like 'aaaa' and 'aaac', regardless of how their sequences change or the diversity of their torsions, the specific three-dimensional shapes they can sample are significantly restricted due to closure constraints.

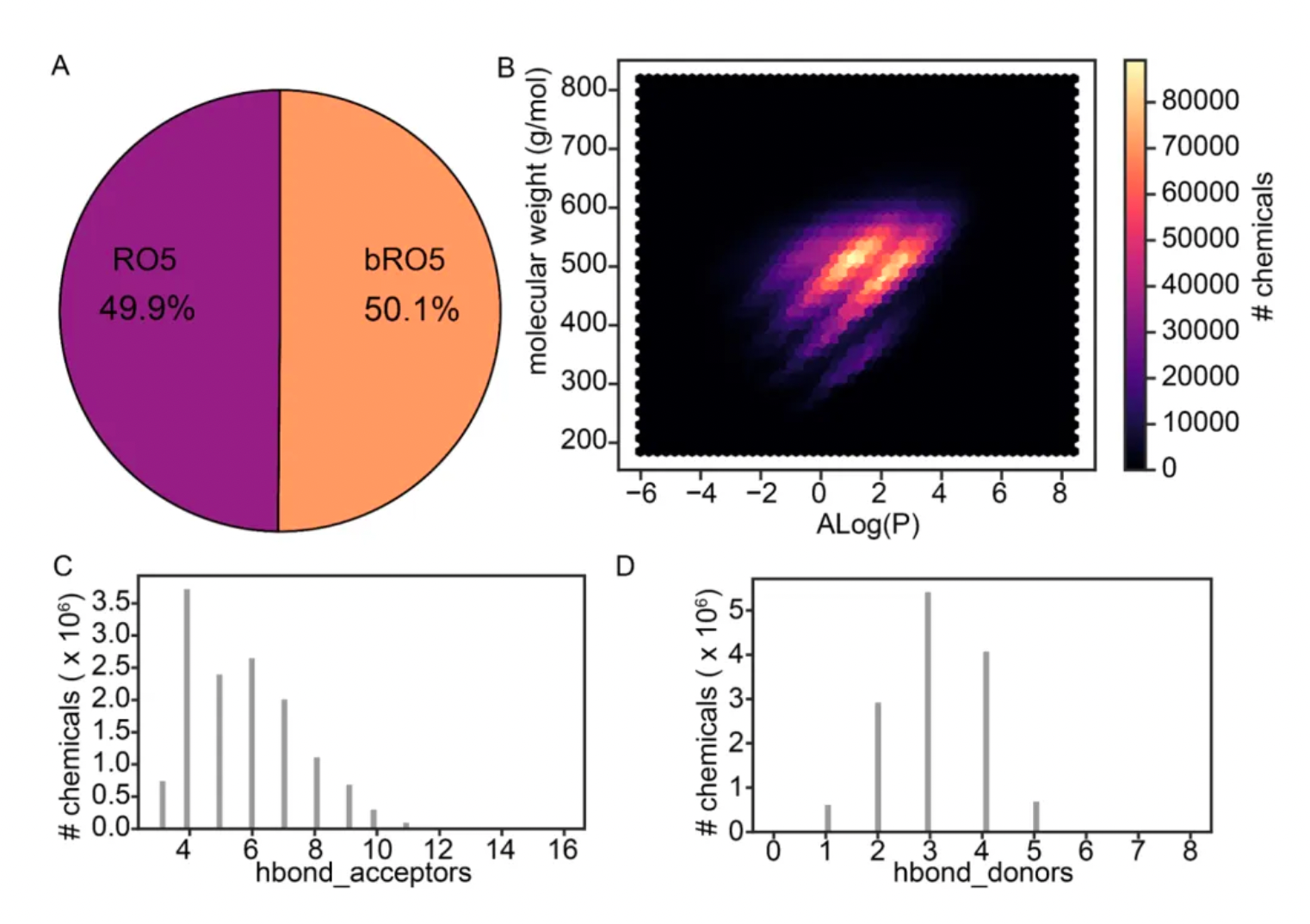
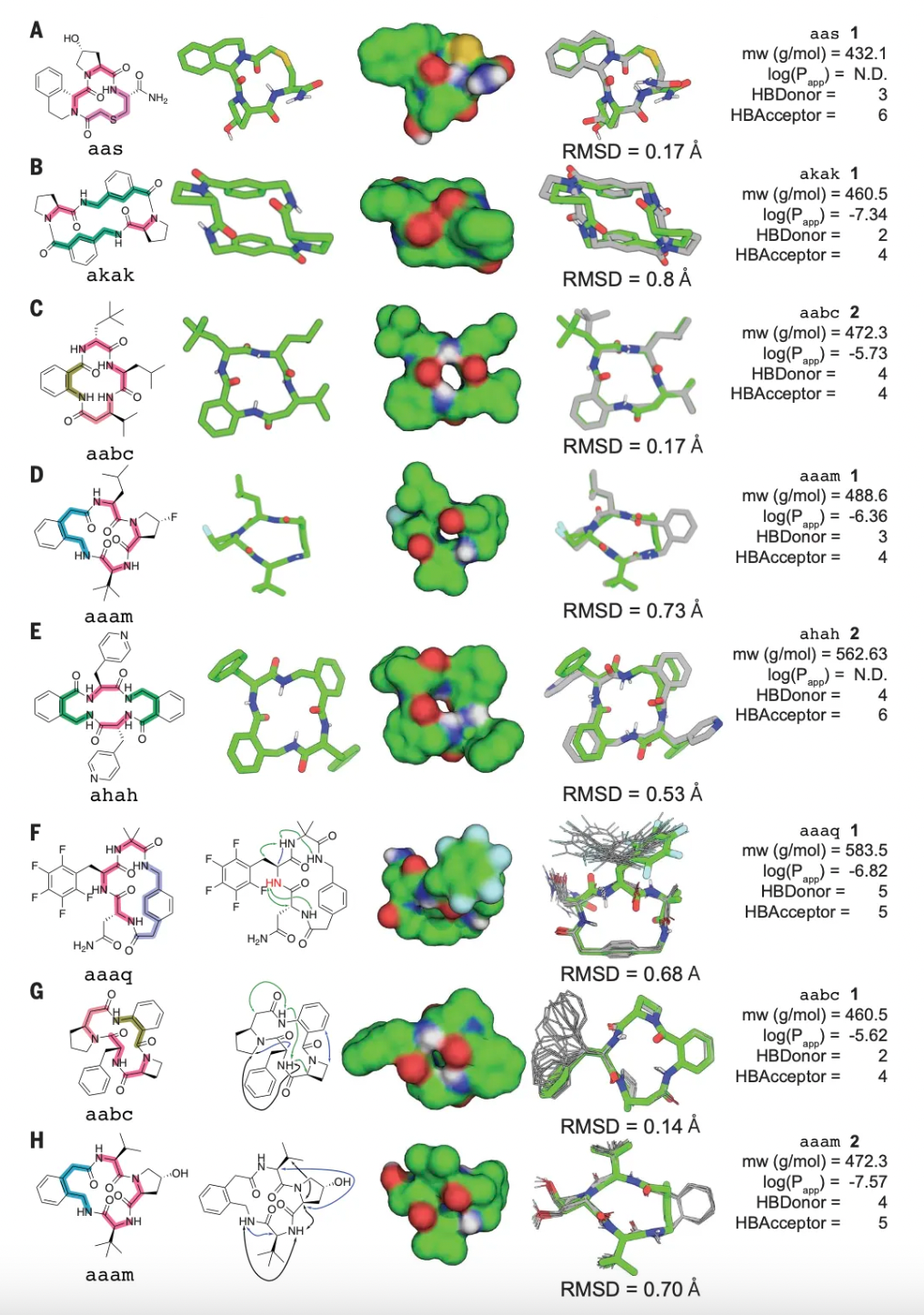
The study shows that about half of the unique macrocycles listed in this research comply with Lipinski's Rule of Five. Macrocycles that adhere to this rule generally exhibit better oral pharmacokinetic properties in drug development, whereas those that violate the rule may face challenges in absorption, distribution, metabolism, and excretion. The corresponding molecular weights, Alog(P), and distributions of hydrogen bond donors and acceptors are illustrated in the figure below.
For each type of monomer sequence, a hash-based closure algorithm constructs a collection of macrocycle structure models made up of low-energy monomer conformations. Assuming sufficient computing power is available, each of these collections can undergo minimization using AIMNet to integrate factors such as inter-monomer interactions and closure stresses, and thereby assess by examining the energy landscape depicted by all low-energy conformations whether the monomer sequence encodes a singular low-energy conformation. However, it is impractical to perform complete energy computations on all 14 million generated macrocycle structure collections. Therefore, the research team focused on two subsets of closed macrocycles considered particularly likely to possess a singular ground state: (i) those whose local interactions strongly favor one or a few closed states; and (ii) those containing non-local hydrogen bonds between main chain amides. The design and investigation of these two types of macrocycles are more likely to efficiently reveal potential drug candidates and advance structure-based drug design efforts.
Research personnel further selected 18 macrocycles predicted to be in a singular low-energy stable state for chemical synthesis, and the precise three-dimensional structures of these compounds were determined using X-ray crystallography or nuclear magnetic resonance techniques. Encouragingly, the experimentally measured structures of 15 of these compounds were in high agreement with the design models, demonstrating the high precision of this method in prediction.
To validate the therapeutic application of these macrocyclic compounds, researchers have developed selective inhibitors targeting three currently prominent protein targets. This not only confirms the immense potential of these novel macrocyclic designs in drug discovery but also signifies a major breakthrough in the field of structure-based drug design.