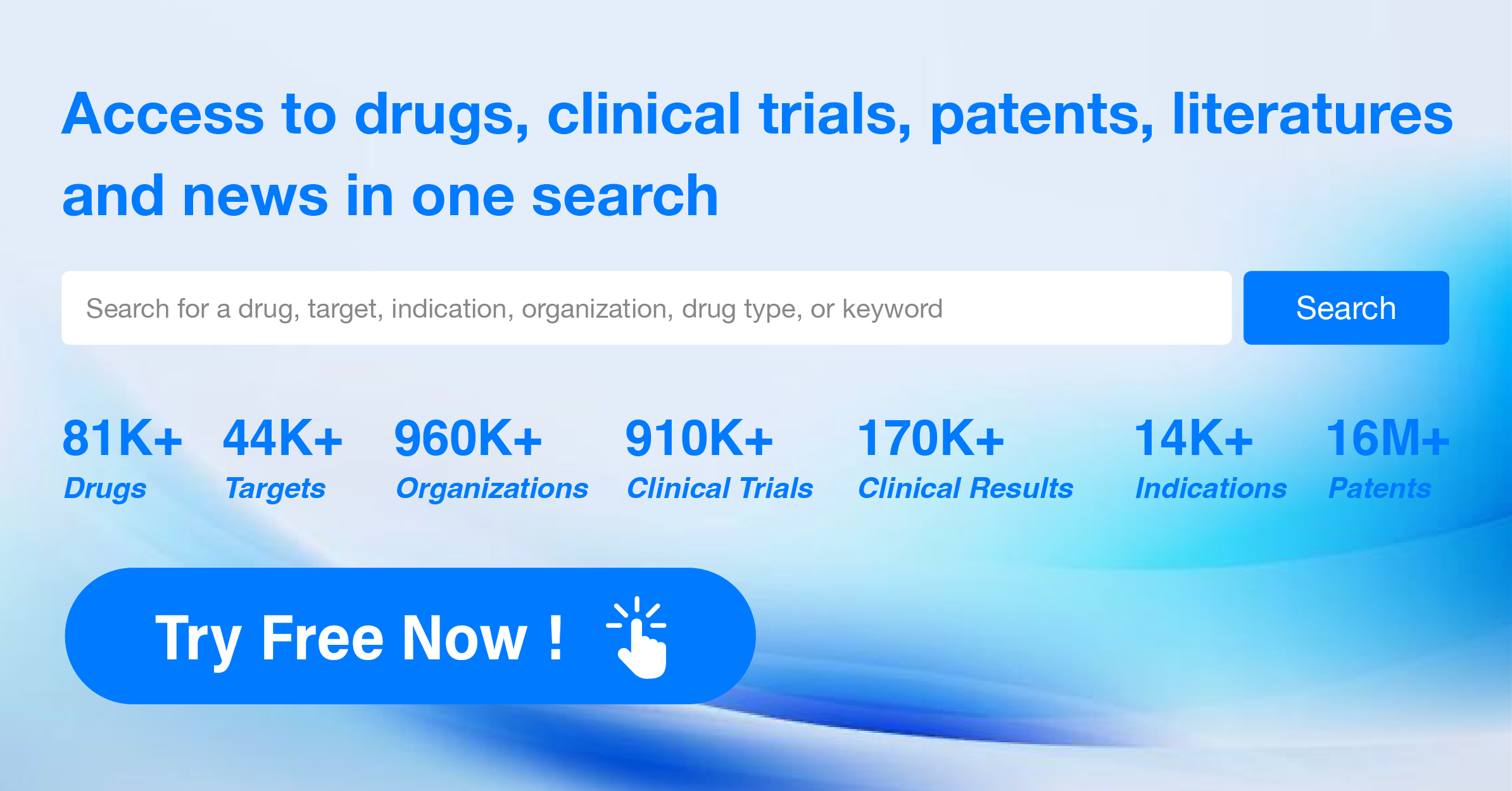
Pharma Frontiers: Daily Digest of Global Pharmaceutical News - April 19
1.Novartis Announces Sustained High Efficacy of CD20-Targeted Therapy Kesimpta for Up to 6 Years
On April 18th, Novartis announced data from its ALITHIOS open-label extension study demonstrating that first-line treatment with Kesimpta (ofatumumab) could maintain high efficacy for up to six years in newly diagnosed patients with relapsing multiple sclerosis (RMS). Patients considered recently diagnosed were defined as those who began treatment within three years of initial diagnosis and had not received any other therapy prior. The efficacy observed included a 44% reduction in the number of relapses in patients who continued with first-line Kesimpta treatment compared to those who later switched to Kesimpta; a reduction of 96.4% and 82.7% in MRI lesions (Gd+ T1 and new T2) respectively; and a reduction of 24.5% and 21.6% in confirmed disability worsening (CDW) events at 3 and 6 months respectively. The analysis showed that the annualized relapse rate (ARR) for newly diagnosed, treatment-naïve RMS patients was further reduced in the ALITHIOS open-label extension study, from 0.104 in a prior Phase 3 clinical study to 0.050, a reduction of 52.0%, equivalent to experiencing a relapse once every 20 years.
Kesimpta is a humanized monoclonal antibody that targets CD20, designed to deplete B-cells from the circulatory system by binding to the CD20 present on their surface. It was approved by the FDA in 2020 for the treatment of adults with relapsing multiple sclerosis. The press release highlighted that Kesimpta is the first B-cell-targeted therapy that allows patients to self-administer at home once a month with the Sensoready autoinjector pen, offering convenience in managing their disease.
2.GSK announces long-term follow-up results for its shingles vaccine, with a protection efficacy of 82% after 10 years!
On April 18th, GSK announced positive long-term data from the phase III clinical trial ZOSTER-049 for its recombinant shingles vaccine, Shingrix (RZV). The trial tracked participants for up to approximately 11 years following vaccination with Shingrix. The final data revealed that RZV provided protection against shingles for over ten years in adults aged 50 and above. The primary outcomes of ZOSTER-049 included: a cumulative vaccine efficacy (VE) of 79.7% (95% CI, 73.7-84.6) between the sixth and eleventh years post-vaccination in adults aged 50 and above. In the eleventh year, the vaccine efficacy in adults aged 50 and above was 82.0% (95% CI, 63.0-92.2), demonstrating that the yearly efficacy of the vaccine remained high post-vaccination. From the sixth to the eleventh year post-vaccination, the cumulative vaccine efficacy in adults aged 70 and above was 73.1% (95% CI, 62.9-80.9), indicating high efficacy across all age groups. During the follow-up period of ZOSTER-049, no new safety concerns were identified. No serious adverse events were considered by the researchers to have a causal relationship with RZV vaccination. In adults aged 50 and above, the most commonly reported adverse reactions to RZV included pain at the injection site, muscle pain, fatigue, and headache, most of which were mild to moderate and typically resolved within three days.
Shingles is caused by the reactivation of the varicella-zoster virus (VZV), which also causes chickenpox. Shingrix is a non-live, recombinant subunit vaccine used to prevent shingles in adults aged 50 and over. It combines the antigen, glycoprotein E, with the adjuvant system AS01B, which helps to overcome immunosenescence related to aging, crucial for protecting individuals aged 50 and over from shingles attacks.
3. GSK's Potential "First-in-Class" Small Molecule Therapy Reaches Key Phase 3 Clinical Endpoint
On April 18th, GSK announced positive results from the pivotal phase 3 clinical trial EAGLE-1 for its potential “first-in-class” oral antibiotic gepotidacin, targeting uncomplicated urogenital gonorrhea in adolescents and adults. Gepotidacin achieved a microbiological cure rate of 92.6%, meeting non-inferiority criteria when compared to the active control medication. The results of the EAGLE-1 trial were based on microbiological response (successful eradication of the bacterial cause of gonorrhea) as the primary endpoint. The trial demonstrated that gepotidacin, compared to the common gonorrhea combination treatment regimen (intramuscular ceftriaxone plus oral azithromycin), met non-inferiority criteria, with a treatment success rate of 92.6%, as opposed to 91.2% in the latter. In the EAGLE-1 trial, the safety and tolerability profile of gepotidacin was consistent with results from phase 1 and 2 trials. The most commonly reported adverse events (AEs) among participants were associated with the gastrointestinal (GI) tract. All adverse events were mild or moderate (Grade 1 or 2).
Gonorrhea is a sexually transmitted infection caused by the bacterium Neisseria gonorrhoeae, which is classified as a priority pathogen by the World Health Organization. It affects both men and women and, if not properly treated, can lead to infertility and other sexual and reproductive health complications. It also increases the risk of HIV infection. Discovered by GSK scientists, gepotidacin is a potential "first-in-class" triazaacenaphthylene antibiotic currently under research. It inhibits bacterial DNA replication through a novel mechanism of action and binding site, providing good balanced inhibition of two different types of Type II topoisomerases in most pathogens.
4. Roche's High-Profile Antibody Therapy Ocrevus Shows Positive Phase 3 Clinical Trial Data, Anticipated Approval This Year
On April 18th, Genentech, a member of the Roche Group, announced that its high-profile antibody therapy Ocrevus (ocrelizumab) achieved positive results in the Phase 3 clinical study OCARINA II for the treatment of relapsing or primary progressive multiple sclerosis (RMS or PPMS). The findings reveal that just two doses of subcutaneous injection of Ocrevus per year can almost completely suppress clinical relapses and brain lesions. Ocrevus is a humanized monoclonal antibody designed to target CD20-positive B cells, which are a specific type of immune cell believed to be a key factor in myelin and axonal damage. The regulatory application for the subcutaneous formulation of Ocrevus is currently under review by regulatory authorities in Europe and the United States, with hopes of approval within this year.
Updated long-term follow-up results indicate that the subcutaneous Ocrevus injection (920 mg; n=236) nearly completely suppressed relapse activity during the treatment phase (97.2% of patients did not experience a relapse). The annualized relapse rate over 48 weeks of Magnetic Resonance Imaging (MRI) monitoring was 0.04, with the vast majority of patients having no Gd+T1 lesions and no new/enlarging T2 lesions. These lesion types are indicators of active inflammation and disease burden, respectively. Additional data continue to confirm that the safety profile of Ocrevus subcutaneous injection is consistent with the established safety profile of Ocrevus intravenous infusion. No new safety signals were found. The most common adverse event in the group receiving subcutaneous Ocrevus was injection reactions (51.5% of all treated patients), including erythema (34.8%), pain (17.2%), swelling (9.4%), and pruritus (5.6%), all of which were mild or moderate in severity and did not lead to discontinuation of treatment.
5. Eli Lilly and Company Announces Positive Phase 3 Trial Results for Heavyweight Treatment of Snoring
On April 18th, Eli Lilly and Company announced the positive results from two Phase 3 clinical trials in the SURMOUNT-OSA program. Analysis indicated that the trial met its primary endpoints and all key secondary endpoints. Compared to placebo, the company’s dual GIP and GLP-1 receptor agonist, tirzepatide, significantly improved the condition of snoring in obese patients with moderate to severe Obstructive Sleep Apnea (OSA), regardless of whether the patients were being treated with a ventilation device. Based on these positive results, Eli Lilly plans to submit relevant regulatory applications.
Medically speaking, frequent snoring can be a sign of OSA. This condition occurs when the upper airway completely or partially collapses during sleep, leading to blocked air passageways, which may result in apneas or hypopneas, potentially accompanied by a drop in blood oxygen saturation or awakenings during sleep. To date, no medication for the treatment of OSA has been approved. SURMOUNT-OSA is a multicenter, randomized, double-blind study designed to investigate the efficacy and safety of weekly subcutaneous injections of tirzepatide compared to placebo in 469 obese adults with moderate to severe OSA. Analyses have shown that both studies reached their primary endpoints. Tirzepatide significantly improved symptoms of sleep-apnea in obese patients, with or without Positive Airway Pressure (PAP) treatment. Study 1 evaluated the effects of tirzepatide in patients not undergoing PAP therapy. The analysis revealed that at 52 weeks, tirzepatide led to an average reduction in the Apnea-Hypopnea Index (AHI) of 27.4 events per hour from baseline, compared to 4.8 events per hour for placebo. In terms of key secondary endpoints, tirzepatide resulted in an average reduction in AHI by 55.0% from baseline, whereas for placebo, the reduction was 5.0%.
6. ImmuneOnco Biopharmaceuticals' next-generation CD47-targeting molecule receives Phase III clinical trial approval for combination therapy
On April 17, ImmuneOnco Biopharmaceuticals announced that its Phase 3 clinical trial for the combination therapy of Timdaracept and the anti-PD-1 monoclonal antibody Tirelizumab has been approved by the Drug Evaluation Center (CDE) of the National Medical Products Administration (NMPA) of China. The indication for this trial is for the treatment of patients with classical Hodgkin’s lymphoma (cHL) who are refractory to anti-PD-(L)1 monoclonal antibody therapy.
Timdaracept, a next-generation CD47-targeting molecule developed by ImmuneOnco, possesses a dual mechanism of action: it can block the tumor's "don't eat me" signal while activating the patient’s immune system's "eat me" signal through IgG1. The product exhibits strong anti-tumor activity in vivo and has demonstrated efficacy as a monotherapy in clinical observations. In preclinical in vivo pharmacodynamic studies, Timdaracept used in combination with targeted drugs or immunotherapeutic agents exhibited potent tumor suppression against both hematological malignancies and solid tumors. In December 2023, the results of a Phase 2 clinical study of Timdaracept for the treatment of cHL following the failure of PD-(L)1 antibody therapy were selected for an oral presentation at the annual meeting of the American Society of Hematology (ASH). According to the updated data presented by ImmuneOnco at the 2023 ASH conference, Timdaracept combined with Tirelizumab achieved an objective response rate (ORR) of 65.2%, with a complete response (CR) rate of 17.4% and a disease control rate (DCR) of 100% for cHL patients who had failed previous anti-PD-(L)1 antibody therapy. Furthermore, subgroup analysis demonstrated that patients could benefit from the combination therapy across various subgroups. Additionally, the tolerance of IMM01 in combination with Tirelizumab was well-received, with no patients discontinuing treatment permanently due to drug-related adverse reactions.
7. Sanofi's Potential "First-in-Class" Monoclonal Antibody Displays Promising Therapeutic Efficacy
On April 18, Sanofi announced the one-year treatment results from a Phase 2 trial of its potential "first-in-class" monoclonal antibody, frexalimab, for the treatment of patients with multiple sclerosis (MS). Analysis data from the trial at 48 weeks indicate that patients treated with frexalimab continued to experience a reduction in disease activity and demonstrated good tolerance. Sanofi has initiated Phase 3 clinical trials of frexalimab for the treatment of relapsing MS and non-relapsing secondary progressive MS. The Phase 2 trial results being reported is a randomized, double-blind, placebo-controlled study that assessed the efficacy of frexalimab in patients with relapsing MS. Patients were randomly assigned (4:4:1:1) to receive either a high dose (1200 mg of frexalimab by intravenous injection every four weeks) or a low dose (300 mg of frexalimab by subcutaneous injection every two weeks) of frexalimab or the matching placebo, for a duration of 12 weeks (Part A). The primary endpoint was the number of new Gd+ T1 lesions on brain MRI at week 12. Secondary endpoints included other MRI-based efficacy measures, as well as the safety, tolerability, and pharmacokinetics of frexalimab. After week 12, participants previously on placebo switched to the corresponding frexalimab group and entered the ongoing open-label extension (OLE) Part B. Frexalimab (SAR441344) is a novel CD40 ligand (CD40L)-targeted monoclonal antibody, believed to interrupt the necessary costimulatory CD40/CD40L pathway for the activation and function of adaptive (T and B cells) and innate (macrophages and dendritic cells) immune cells, without necessitating lymphocyte depletion.
8. InnoCare Pharma Announces Phase 2 Study Data of BTK Inhibitor for the Treatment of Autoimmune Diseases
On April 18th, InnoCare Pharma announced that the results of its Phase 2 clinical study on the Bruton's tyrosine kinase (BTK) inhibitor Orelabrutinib for the treatment of patients with chronic immune thrombocytopenia (ITP) have been recently published in the American Journal of Hematology. The study indicates that Orelabrutinib could provide a safe and effective treatment option for patients with ITP. As outlined in an InnoCare Pharma press release, a Phase 3 registrational clinical study of Orelabrutinib for ITP patients is progressing rapidly with patient enrollment expected to be completed by the end of 2024. ITP is a rare and severe autoimmune disease characterized by a low platelet count in the blood, which can lead to serious bleeding events. In patients with ITP, the immune system attacks and destroys the body's own platelets, which play an active role in blood clotting and wound healing.
Orelabrutinib is a highly selective novel BTK inhibitor developed by InnoCare Pharma for the treatment of hematological malignancies and autoimmune diseases. Currently, Orelabrutinib has been approved in China for three lymphoma indications and has also been approved for marginal zone lymphoma in Singapore. The study published in the American Journal of Hematology aimed to evaluate the efficacy and safety of Orelabrutinib in the treatment of adult patients with chronic ITP. The primary endpoint of the study was the proportion of patients whose platelet count reached at least 50 × 10^9/L for at least two consecutive weeks without the use of rescue medication during the previous four weeks. A total of 33 patients were enrolled in the study, and Orelabrutinib demonstrated good safety in two dose groups of 50 mg and 30 mg administered orally once daily. The once-daily 50 mg dose group showed rapid onset of action and better efficacy, particularly in patients who had responded to previous treatment with glucocorticoids (GC) or intravenous immunoglobulin (IVIG).
9.Genescience Pharmaceuticals has submitted a marketing application for Triptorelin, a gonadotropin-releasing hormone agonist, in the form of microspheres
On April 18th, the official website of the Center for Drug Evaluation (CDE) of the National Medical Products Administration (NMPA) of China publicized that Genescience Pharmaceuticals has submitted a New Drug Application for Triptorelin Acetate Microspheres for Injection, which has been accepted. Public information indicates that Triptorelin is a gonadotropin-releasing hormone (GnRH) agonist and has previously completed a phase III clinical trial for the treatment of central precocious puberty in children.
Triptorelin is an analog of natural GnRH, which stimulates the pituitary gland to periodically secrete gonadotropins, thereby promoting the secretion of sex hormones by the gonads. Sex hormones are related to the development of various diseases, such as endometriosis, uterine leiomyomas (fibroids), breast cancer, prostate cancer, and others. In clinical practice, GnRH agonists have been widely used to treat conditions such as prostate cancer, endometriosis (stages I to IV), female infertility, pre-operative treatment of uterine fibroids, and precocious puberty, among others. According to the official website of the Chinese Clinical Drug Trial Registry and Information Publicity Platform, Genescience Pharmaceuticals has previously completed a multicenter, open-label, single-arm phase III clinical study aimed at evaluating the safety and effectiveness of Triptorelin Acetate Microspheres (Gensci006) in treating central precocious puberty in children.