Pharvaris is developing a bradykinin B2 receptor antagonist for the treatment of hereditary angioedema
Hereditary Angioedema (HAE) is a rare genetic disorder characterized by recurrent episodes of localized subcutaneous and mucosal edema. Patients with HAE have reduced levels of an important protein, C1 Inhibitor (C1-INH), in their blood, or this C1-INH is nonfunctional. These forms of hereditary angioedema are distinct from allergic angioedema, which is a type of skin reaction typically associated with hives (urticaria). Hereditary Angioedema is a potentially life-threatening condition caused by a genetic defect.
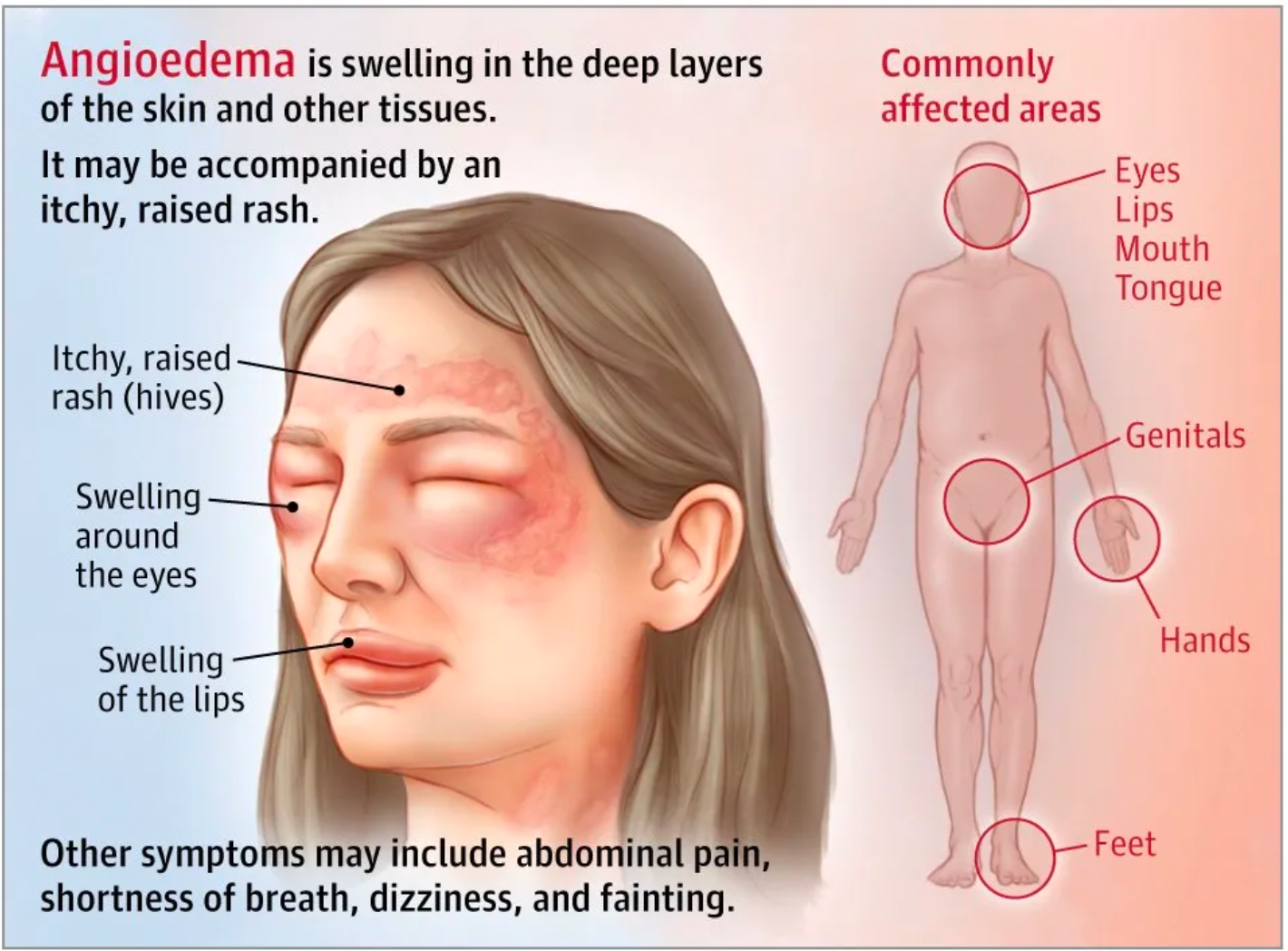
Hereditary Angioedema can cause painful swelling, usually occurring in the face, hands, feet, or genitals. Dangerous swelling can also occur in the airways of the lungs or the walls of the intestines. In the same episode, these symptoms can migrate from one location to another. Most attacks are unpredictable, and the triggering factors vary from person to person. Potential triggers may include stress, surgical procedures, dental work, medications, and illnesses such as the common cold. Many individuals have early warning signs of an HAE attack, such as extreme fatigue, skin tingling, hoarseness, or sudden mood changes that may herald an impending attack. Accurately diagnosing HAE can be difficult because the disease is very rare, and doctors may initially rule out more common diseases with similar symptoms. Besides physical examination and medical history, HAE can also be diagnosed by measuring the level and function of C1-INH in the blood.
As early as the 19th century, scientists first described the clinical manifestations of this disorder and classified it as an autosomal dominant genetic disease. In the past 40 years, scientific research has identified the fundamental defect in hereditary angioedema as a deficiency of functional C1-INH protein and identified bradykinin as the biological mediator of the edema. In hereditary angioedema with C1-INH deficiency, the activation of the plasma contact system produces bradykinin, which exerts its biological effects through the interaction with bradykinin B2 receptor, a member of the G protein-coupled receptor family.
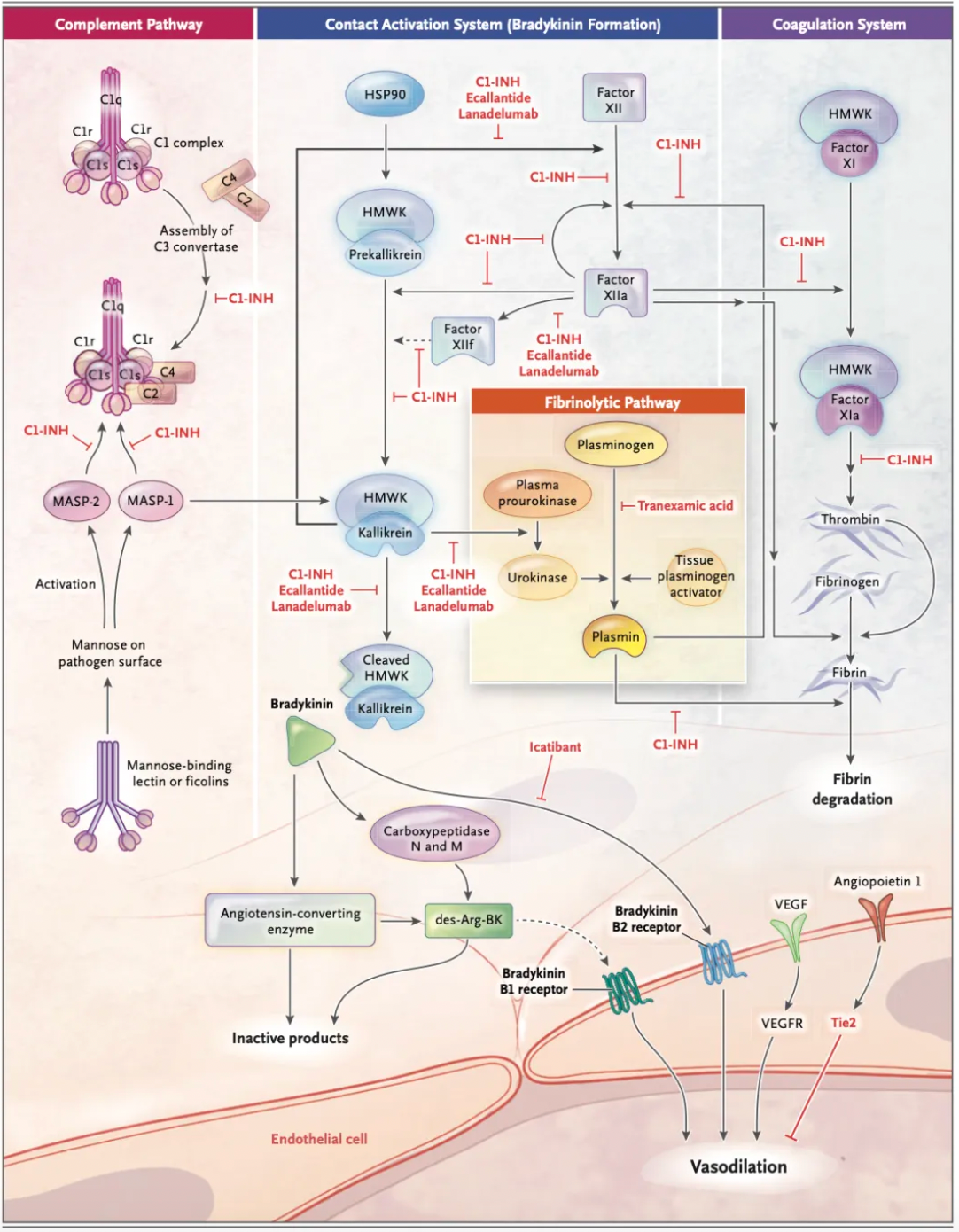
The bradykinin B2 receptor is continuously expressed on endothelial cells and is thought to be primarily responsible for actively transporting fluid into local tissues, leading to vascular edema. Mechanistically, activation of the bradykinin B2 receptor leads to the disassembly of adherens junctions, which play a crucial role in limiting vascular permeability. Following the involvement of the bradykinin B2 receptors, downstream signaling results in the phosphorylation of transmembrane vascular endothelial adhesion molecules, leading to their internalization and degradation. The subsequent contraction of the actin cytoskeleton increases the size of the gaps between endothelial cells, causing vascular leakage.
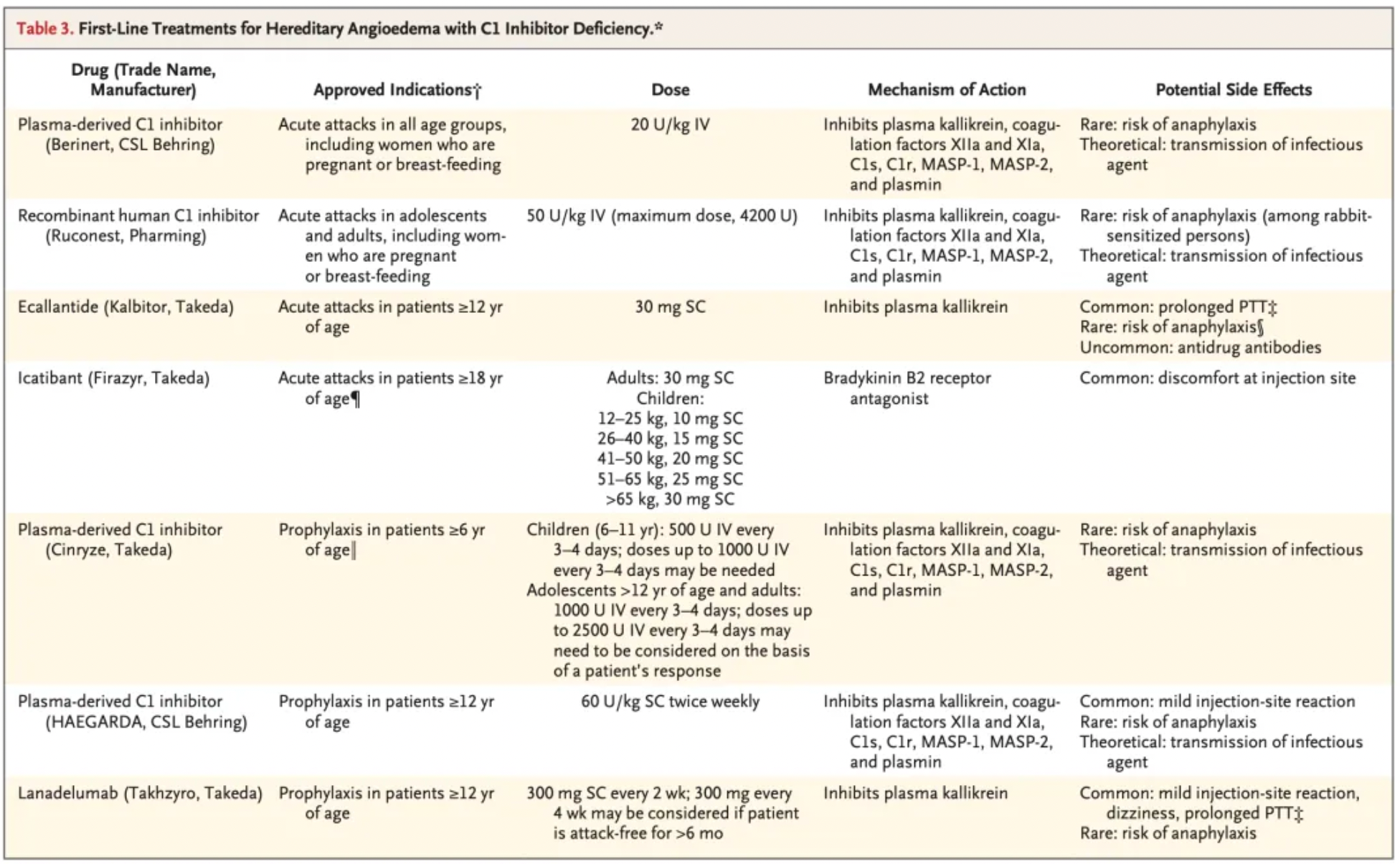
The figure below summarizes the first-line treatment methods for hereditary angioedema (HAE) due to C1-INH deficiency, including the blood-derived product Berinert developed by CSL Behring, approved in 1986; recombinant C1-INH developed by Pharming, approved in 2010; recombinant C1-INH by Takeda Pharmaceuticals, approved in 2008; the recombinant peptide Ecallantide, approved in 2009; the small molecule compound Icatibant, approved in 2008; and monoclonal antibody Lanadelumab, approved in 2018.
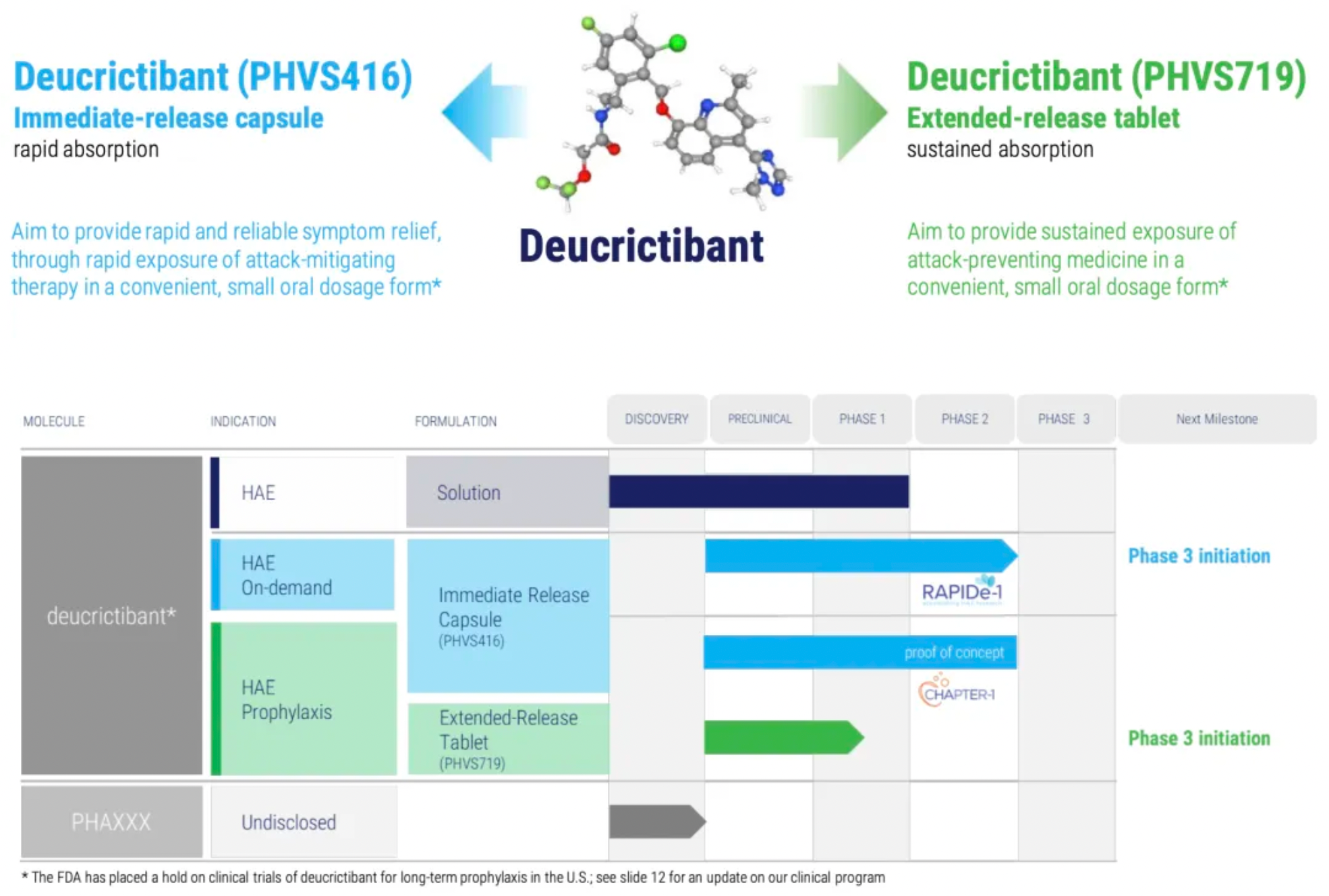
About Pharvaris and Deucrictibant
Pharvaris is a clinical-stage company, leveraging its profound expertise in the field of hereditary angioedema (HAE) to develop novel oral bradykinin B2 receptor antagonists for treating and preventing HAE attacks. Pharvaris aims to provide a safe, effective, and convenient treatment option for patients with all subtypes of HAE by utilizing novel small molecules targeting this clinically validated therapeutic goal, enabling both on-demand treatment and prophylactic management of attacks.
Deucrictibant is a new, potent oral small-molecule bradykinin B2 receptor antagonist, originally developed by the Pharvaris team. The company is currently developing two oral formulations with the same active ingredient - Deucrictibant (PHVS416 and PHVS719) - for on-demand treatment and prophylaxis of hereditary angioedema (HAE).
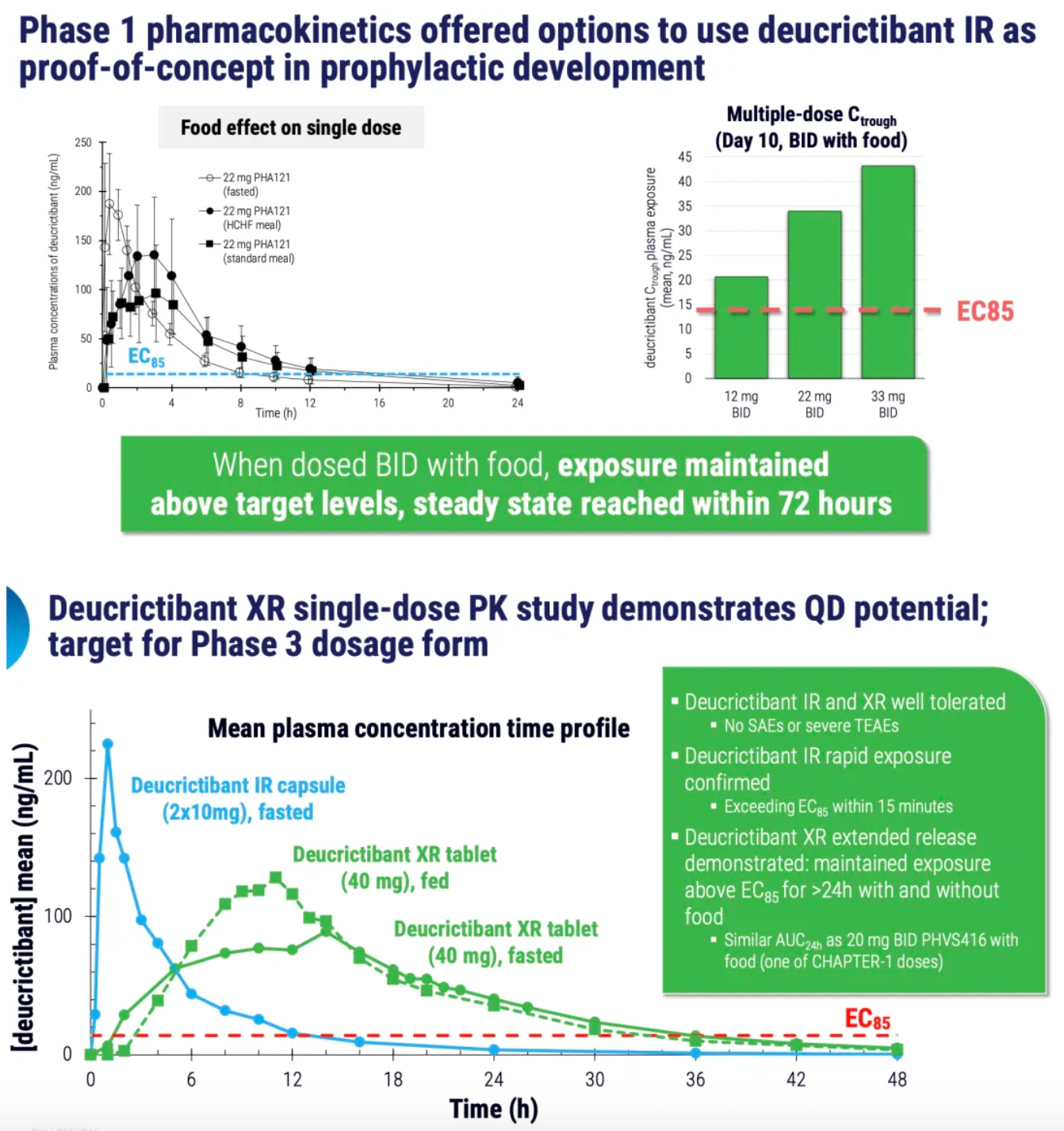
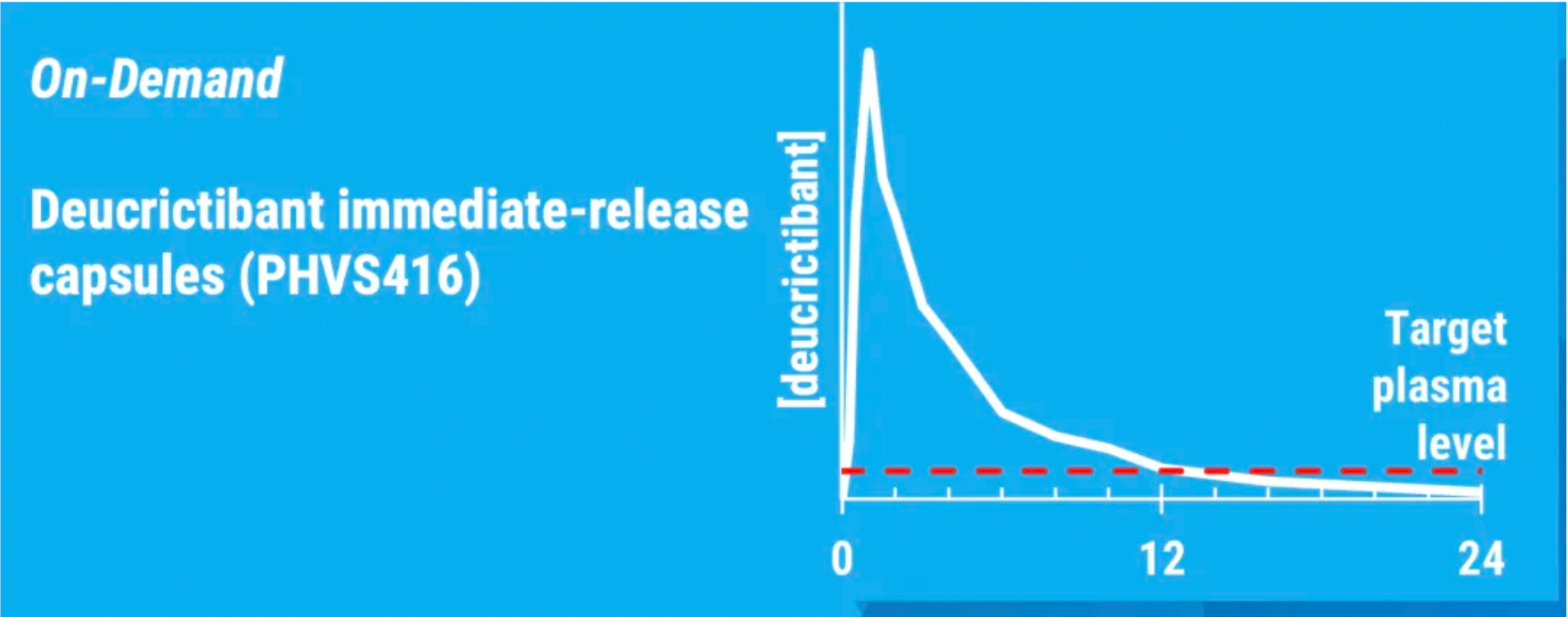
PHVS416 is an experimental soft capsule formulation containing Deucrictibant. PHVS416 is designed as a convenient, compact oral dosage form that alleviates symptoms quickly and reliably through a rapid onset of action for attack relief. In a phase 2 study, PHVS416 consistently demonstrated positive effects across all endpoints and evaluations, confirming the efficacy and tolerability of PHVS416 in the treatment of HAE attacks. Currently, PHVS416 is undergoing phase 2 clinical development outside the United States for both on-demand and prophylactic concept-validation treatment of HAE.
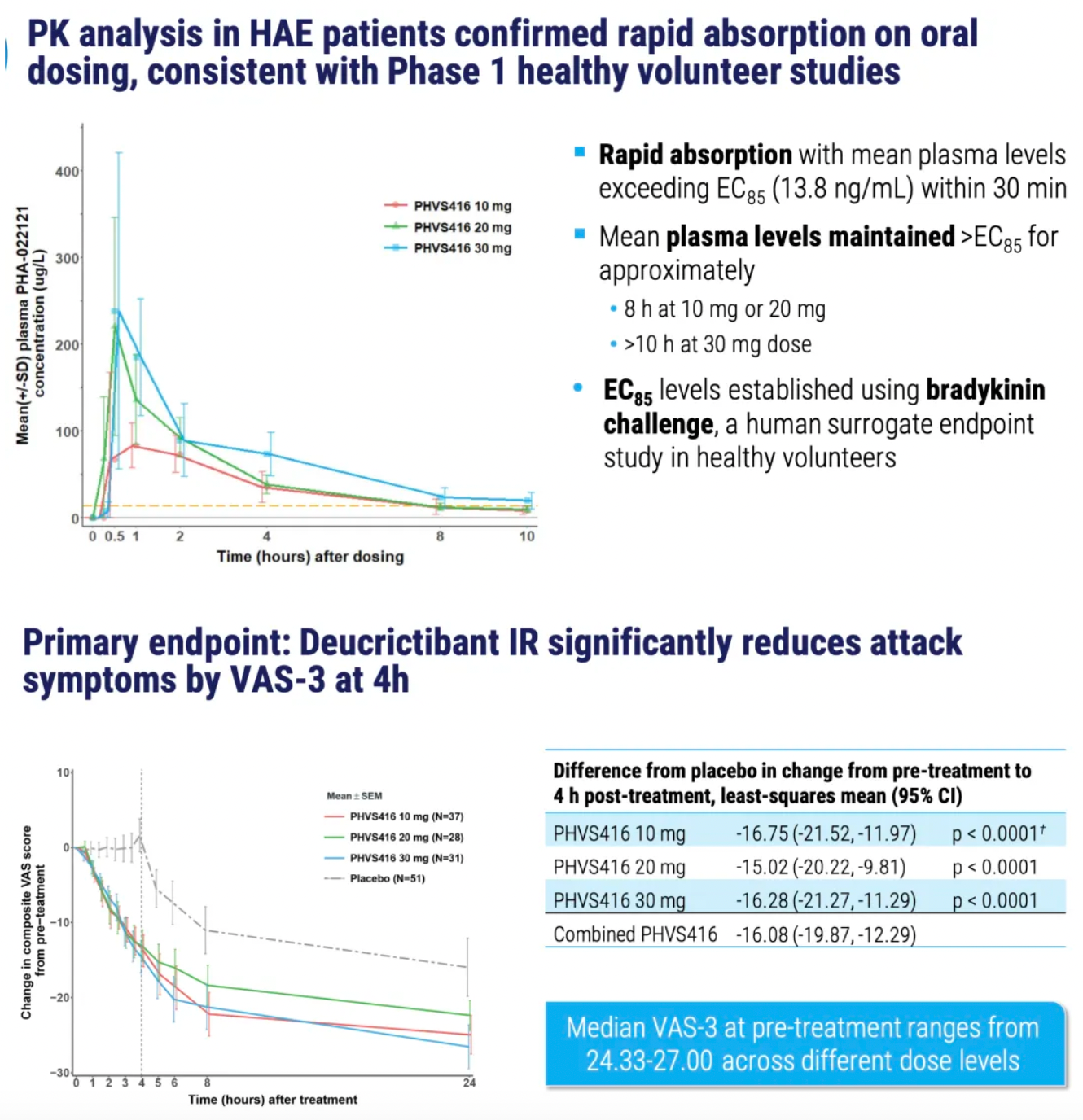
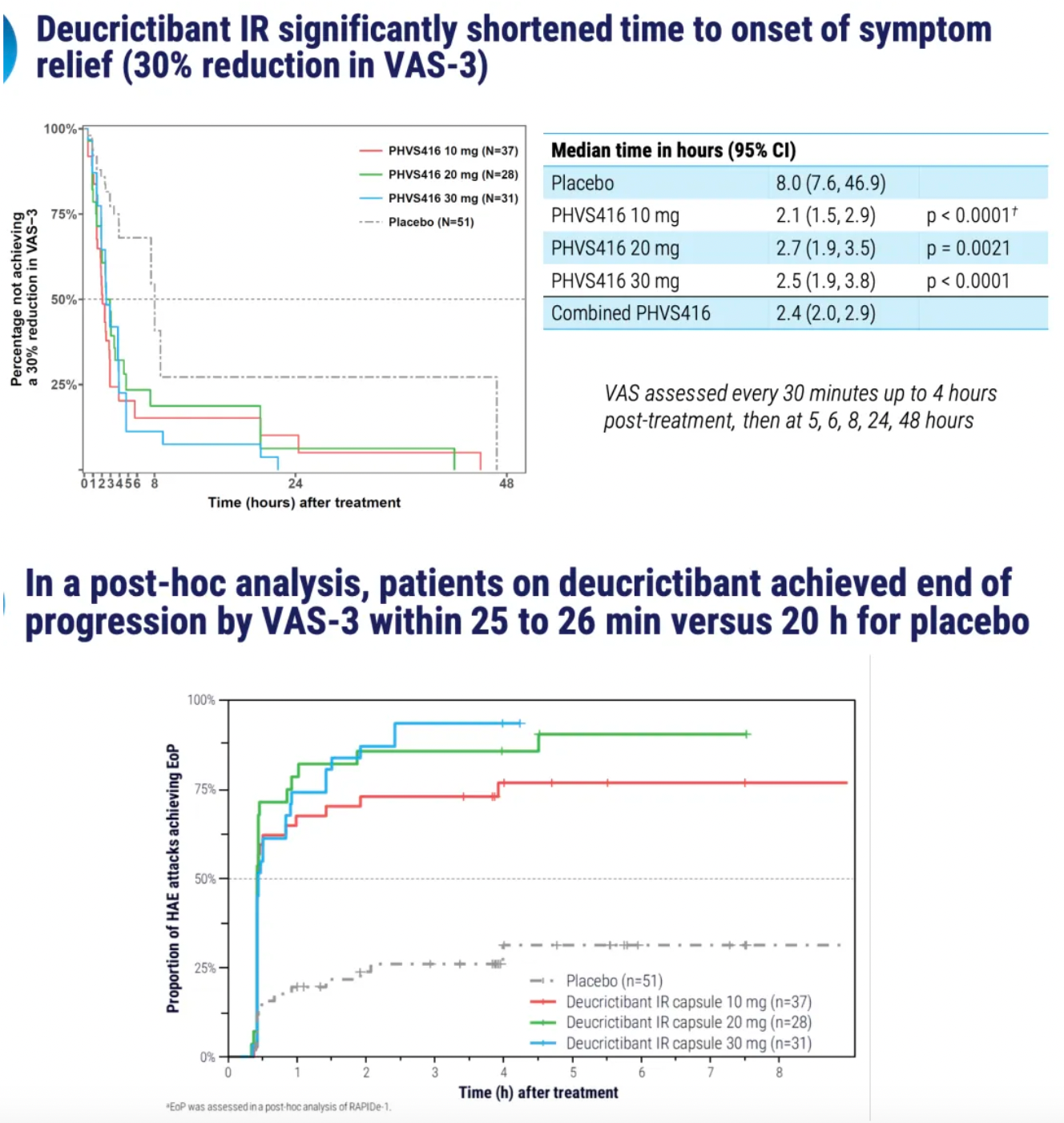
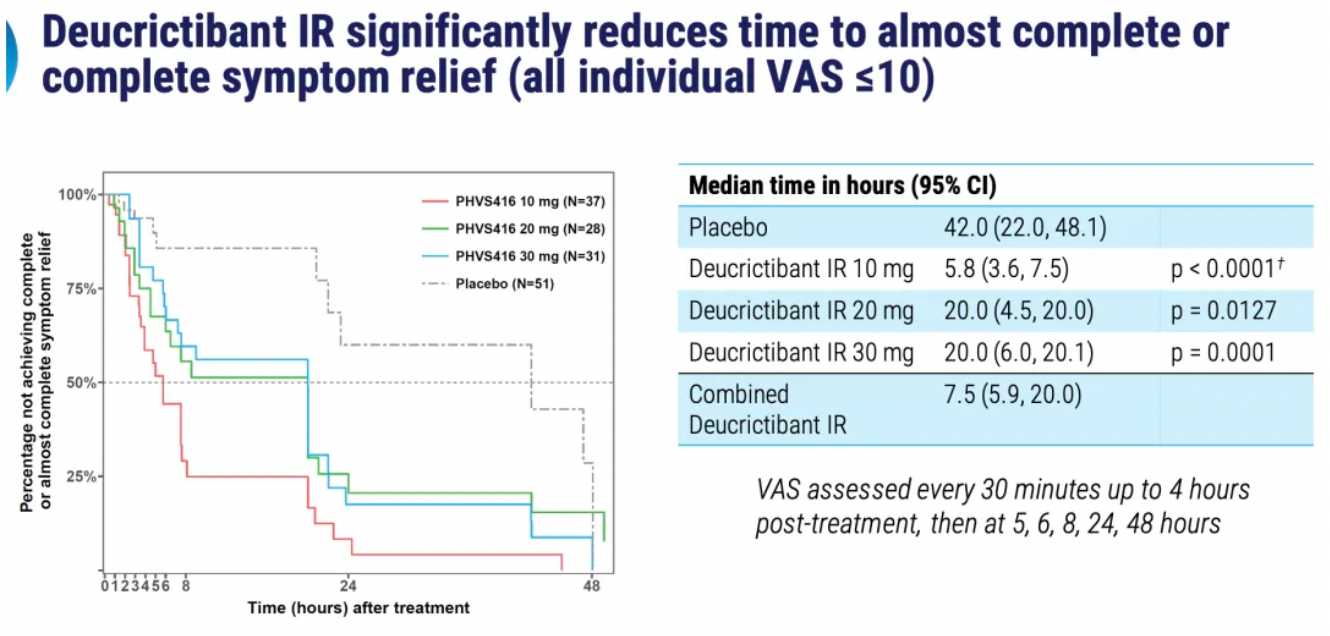
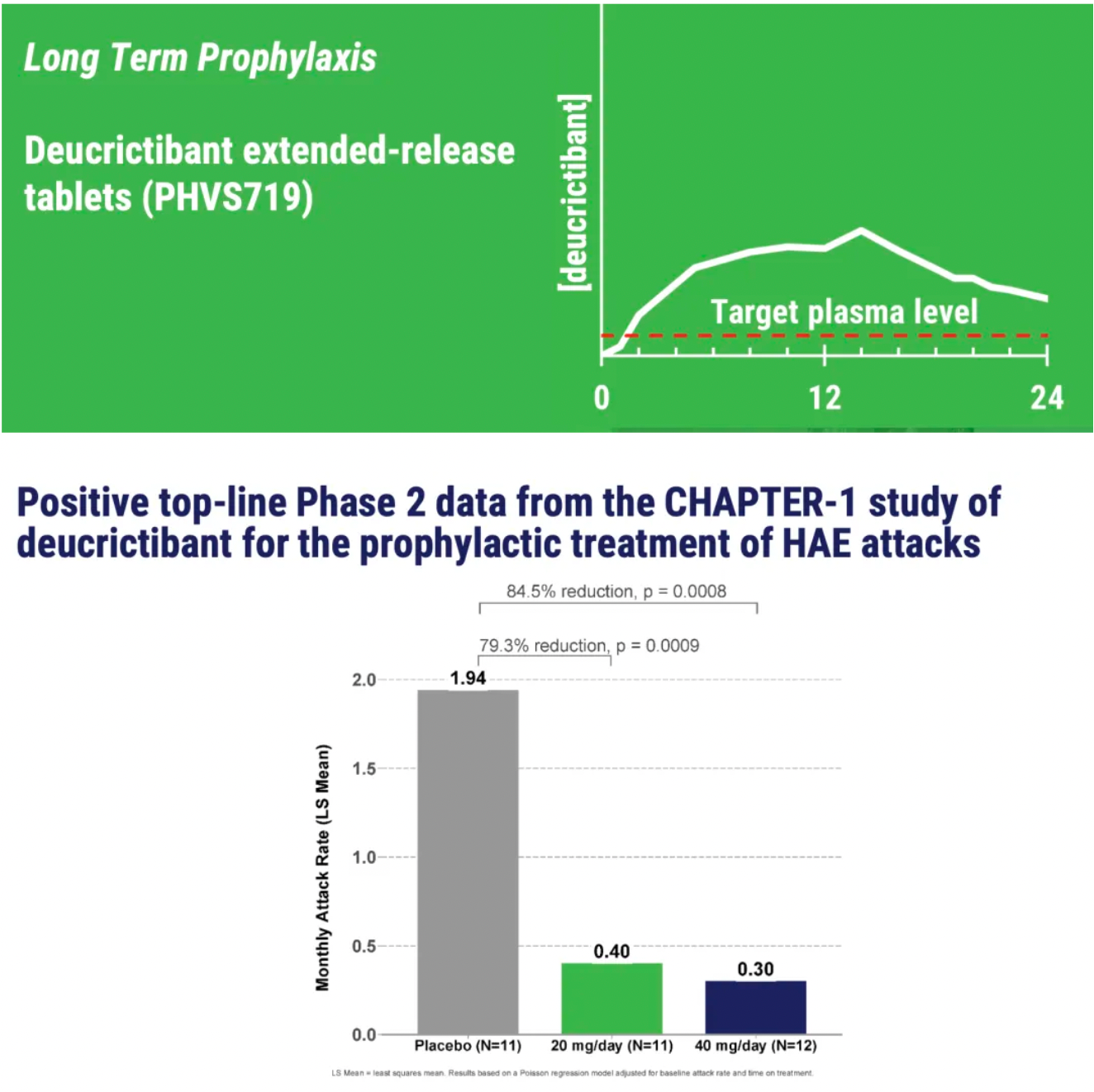
PHVS719 is an experimental sustained-release tablet containing Deucrictibant. PHVS719 is designed for the prevention of HAE attacks, with a conveniently small oral dosage form. PHVS719 is currently in Phase 1 clinical development for prophylactic treatment of HAE. In healthy volunteers, PHVS719 demonstrated good tolerability with a single dose, and its sustained-release properties support once-daily dosing.