AXS-05 or Ketamine,Which is a better choice for depression?
The classification of antidepressants can roughly be divided into several categories: Selective Serotonin Reuptake Inhibitors (SSRIs), Serotonin and Norepinephrine Reuptake Inhibitors (SNRIs), Noradrenergic and Specific Serotonergic Antidepressants (NaSSAs), Norepinephrine and Dopamine Reuptake Inhibitors (NDRI), Melatonin Receptor Agonists, etc. which belong to the first line of treatment. While Serotonin Antagonists and Reuptake Inhibitors (SARIs), Noradrenaline Reuptake Inhibitors (NARIs), Selective Serotonin Reuptake Accelerators (SSRAs), Tricyclic Antidepressants (TCAs), etc. are considered second-line treatments. However, the current antidepressants often have delayed treatment effects and are accompanied by severe side effects. For most patients with severe depression, monoamine-targeted first or second-line treatments often cannot achieve sufficient therapeutic effects.
In 2019, the US FDA approved the use of esketamine for the treatment of treatment-resistant depression. Esketamine shows a rapid and potent antidepressant effect, giving patients new hope and leading to the demand for rapid-acting and alternative mechanism drugs. On August 19, 2022, AXS-05 was also approved for marketing. As a drug of the same class, it has some advantages over esketamine. Dextromethorphan has pharmacological properties similar to esketamine, and when used in combination with bupropion, they have synergistic therapeutic effects. Bupropion works by binding to the neurotransmitter transporters NET and DAT and also has an inhibitory effect on CYP450 2D6, which can improve the oral bioavailability of dextromethorphan.
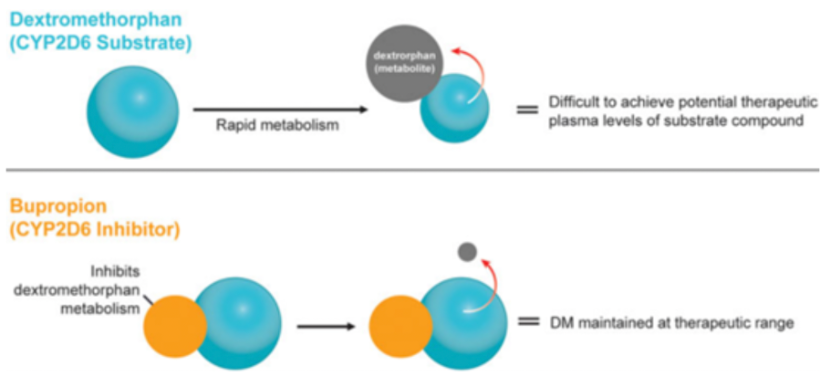
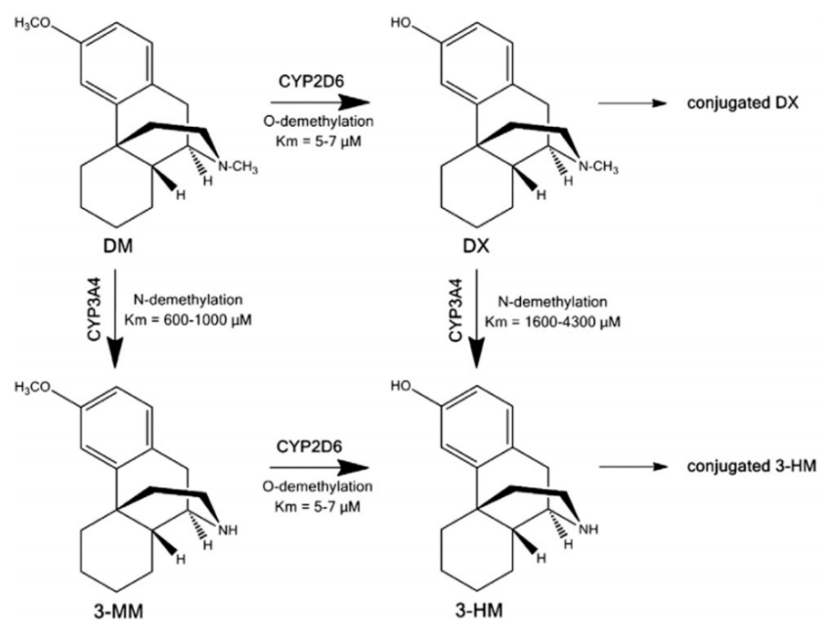
AXS-05 is a combination drug composed of 45mg of dextromethorphan and 105mg of bupropion. Dextromethorphan is primarily considered as an NMDA antagonist, whereas bupropion is a norepinephrine-dopamine reuptake inhibitor. Both bupropion and its metabolites fall under the category of CYP2D6 inhibitors, while dextromethorphan is a substrate of the CYP2D6 metabolic enzyme. Based on the data in Figure 2, bupropion, by inhibiting the CYP2D6 metabolizing enzyme, can reduce dextromethorphan's demethylation metabolic pathways, thus lowering the concentration of the main metabolic product 3-HM, which doesn't have pharmacological activity. At the same time, this also decreases the phase II metabolism of dextromethorphan. In clinical research, the combined use of bupropion and dextromethorphan has indeed significantly increased the plasma exposure of dextromethorphan in all evaluated dose groups. Bupropion possesses a unique central mechanism of action, serving not only as a metabolic inhibitor of dextromethorphan, but also showing pharmacological synergy with dextromethorphan in a wide range of neuropsychiatric diseases (such as depression). Apart from gaining approval for major depressive disorder, AXS-05 is also undergoing phase III (agitation) and phase II (smoking cessation) clinical studies for two other indications.
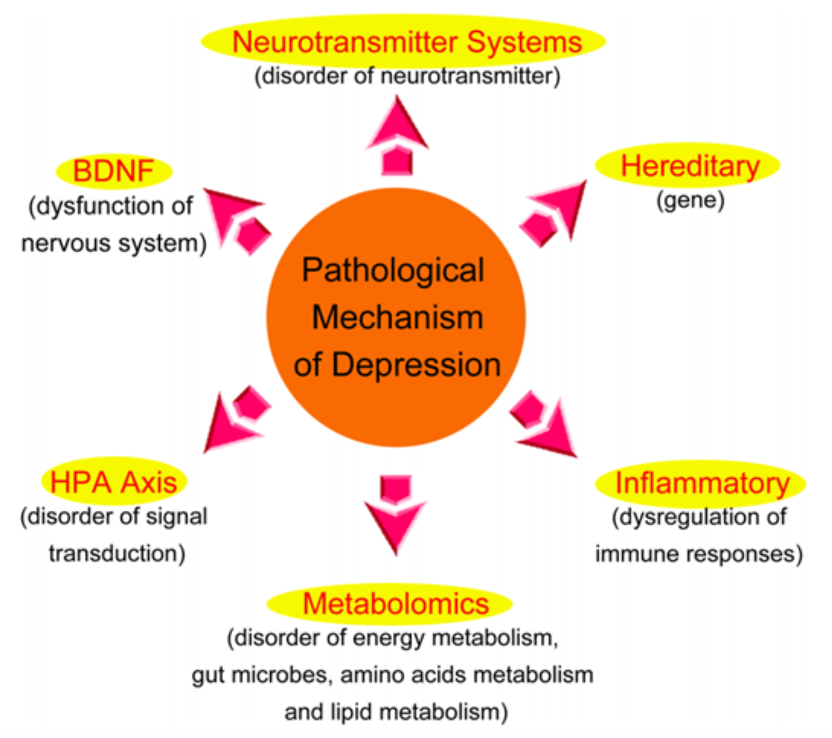
Depression is a prevalent chronic and severe mental disorder, characterized primarily by persistent low mood, sleep disturbance, appetite disorders, and social phobia, among others. Globally, over 300 million people suffer from depression, and nearly 800,000 succumb to suicide due to the condition annually. Depression represents about 10% of the total global burden of non-fatal diseases, resulting in a higher number of years lost to disability than any other disease. Therefore, depression has massive adverse impacts on human health and the economy.
Pathogenesis of Depression:
1. Genetic Factors: The genetic probability of severe depression is likely between 31% and 42%, and the genetic level of recurrent severe depression may be even higher.
2. Neurotransmitter System: The monoamine hypothesis suggests that the reduction in the concentration of monoamine neurotransmitters such as 5-hydroxytryptamine (5-HT) and dopamine (DA) is one of the main causes of depression.
3. Neurotrophic Factor Hypothesis: This hypothesis suggests that alterations in neurotrophic factors, especially brain-derived neurotrophic factor (BDNF), whose expression and function are reduced, might cause emotional depression and hence lead to depression. BDNF is an important neurotrophic factor in the central nervous system and plays a key role in the growth and survival of neurons. Studies have found that BDNF levels in the hippocampus of suicide victims with depression are significantly decreased, but BDNF expression in the brain areas of suicidal patients undergoing antidepressant treatment significantly increased.
4. Hypothalamic-Pituitary-Adrenal axis (HPA): The HPA axis is an important endocrine system that can maintain homeostasis and stress responses, and has significant regulatory functions. It controls the secretion of various regulatory peptides and hormones and plays a unique role in the onset and development of depression. Clinical studies found that patients with depression often exhibit hyperactivity of the HPA axis.
5. Inflammatory Factors: Patients with depression often have elevated levels of inflammatory factors such as tumor necrosis factor-α (TNF-α) and reduced levels of anti-inflammatory factors. After undergoing antidepressant treatment, the levels of inflammatory factors in the patient's body significantly decrease while the levels of anti-inflammatory factors significantly increase.
6. Excitatory Amino Acid Hypothesis: The excitatory amino acid system, especially the glutamate receptor system and γ-aminobutyric acid receptor system, may be involved in the pathogenesis of depression. Glutamate is an important excitatory amino acid in the central nervous system and is also a vital neurotransmitter in the hippocampal area. Studies indicate that the level of glutamate in the plasma of patients with depression is significantly elevated, and the level of glutamate in cerebrospinal fluid is also significantly elevated. The potential pathogenesis may be that elevated glutamate levels activate N-methyl-D-aspartate receptors (NMDA) on the neuronal cell membrane, leading to the opening of calcium ion channels, a massive influx of calcium ions, and resulting in intracellular calcium overload that leads to neuronal degeneration, or even death.
Research on antidepressant-related drugs has made rapid progress in recent years, with in-depth studies being conducted on the onset mechanisms of depression and specific therapeutic targets. Based on different structural types, antidepressants can be broadly categorized into several types: Selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors, Noradrenergic and specific serotonergic antidepressants (NaSSAs), Norepinephrine-dopamine reuptake inhibitors (NDRIs), Melatonin receptor agonists, etc., which are first-line drugs, as well as Serotonin antagonist and reuptake inhibitors (SARIs), Norepinephrine reuptake inhibitors (NARIs), selective serotonin reuptake accelerators (SSRAs), Tricyclic antidepressants (TCAs), among others, which are second-line drugs.
The classic monoamine hypothesis of depression suggests that depression is caused by a deficiency in monoamine neurotransmitters. Although there is a lack of direct evidence to support the monoamine hypothesis, almost all currently existing antidepressants directly affect one or more monoamine neurotransmitter systems. However, even though these types of drugs increase monoamine levels shortly after treatment begins, the therapeutic effect often takes several weeks to appear.
Therefore, the focus of research has shifted from the neurotransmitters themselves to the receptors and the downstream signals triggered by these receptors, in order to explain the mechanism of action of antidepressants. Increasing attention has been paid to changes in neurogenesis and neuroplasticity mediated by antidepressants. Treatments targeting the monoamine pathway, such as selective serotonin reuptake inhibitors (SSRIs), have been shown to increase neurogenesis in the hippocampal dentate gyrus. These effects may be mediated by the activation of cyclic adenosine monophosphate (cAMP) response element binding protein (CREB), which can regulate the expression of many genes related to neuroplasticity, including brain-derived neurotrophic factor (BDNF). Clinical studies have shown that long-term antidepressant treatment can restore abnormally low BDNF levels, and this recovery is related to a decrease in depression scores.
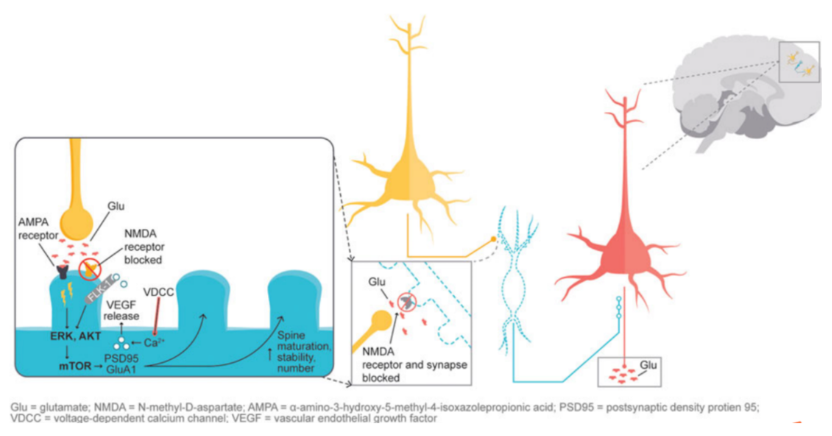
Furthermore, in the previously mentioned excitatory amino acid hypothesis, glutamate signaling is thought to play a crucial role in maintaining neuronal plasticity. When glutamate-mediated AMPA receptors are activated, it causes the activation of extracellular signal-regulated kinase/protein kinase B or ERK/AKT signaling cascade, thereby triggering the activation of the mTOR pathway. The activation of the mTOR pathway can increase the expression of synaptic proteins and subsequently increase the density of dendritic spines. Studies on synaptic and intracellular events stimulated by ketamine show that NMDA antagonism leads to a "glutamate surge", which then increases the flux of AMPA/NMDA receptors, thereby stimulating the intracellular signaling cascade. Studies have shown that both ketamine and dextromethorphan require the activation of AMPA receptors to exert their antidepressant effects. Figure 4 illustrates the downstream cascade reactions involved in neuronal plasticity as induced by the activation of AMPA receptors mediated by NMDA blockade, which is considered to be the basis for antidepressant effects.
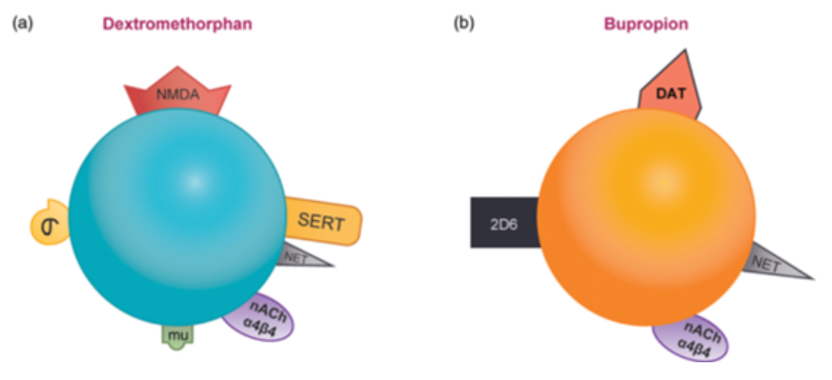
Dextromethorphan/bupropion combines several different mechanisms of antidepressant treatments into one therapy. Both dextromethorphan and bupropion increase the concentration of norepinephrine by inhibiting reuptake, and both are antagonists of α4β4 nACh. In addition, bupropion increases the concentration of dopamine by blocking reuptake, while dextromethorphan increases the concentration of glutamate by acting as an NMDA receptor antagonist and increasing 5-hydroxytryptamine. Dextromethorphan (Figure 5A) is an NMDA receptor antagonist, also an inhibitor of SERT and NET, an antagonist of nACh α4β4, and an agonist of sigma-1 and mu opioid receptors. Bupropion (Figure 5B) is an inhibitor of CYP2D6, also an inhibitor of NET and DAT, and an antagonist of α4β4 nicotinic acetylcholine receptors.
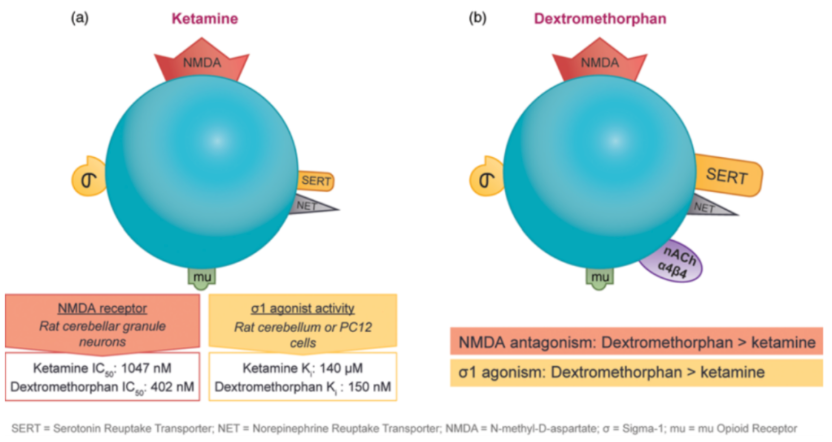
Sub-anesthetic doses of ketamine have an immediate antidepressant effect on patients with treatment-resistant unipolar or bipolar depression, which has triggered investigations into drugs with similar mechanisms of action. Ketamine and dextromethorphan exhibit activity at the same receptor and in some cell test systems, dextromethorphan demonstrates higher affinity as an NMDA receptor antagonist and stronger sigma-1 receptor agonistic action. As shown in Figure 6 (a), the IC50 of ketamine as an NMDA receptor antagonist is 1047 nM, while that of dextromethorphan is 402 nM, an increase of 2.5 times. As for the sigma-1 receptor agonistic effect, the Ki of ketamine is 140 μM, while that of dextromethorphan is 150 nM, an improvement of nearly 1000 times.
Interestingly, before intravenous injection of ketamine to adults with treatment-resistant depression, the opioid antagonist effect of naltrexone attenuated the antidepressant effect of ketamine (but not its dissociative effect). This suggests that the acute antidepressant effect of ketamine requires opioid activation, but not the dissociative effect. One explanation could be that crosstalk between opioid receptor-mediated signaling and NMDA receptor-mediated signaling interferes with ketamine's ability to activate mTOR. However, using the mTOR inhibitor rapamycin before ketamine infusion enhances the treatment of patients and does not block the antidepressant effect of ketamine. Therefore, further research is needed to fully clarify the role of opioid signaling pathways in the antidepressant effect of ketamine.
It is worth noting that AXS-05, composed of dextromethorphan and a CYP2D6 metabolic enzyme inhibitor, is not the first to market. AVP-923, a combination drug with dextromethorphan / quinidine as the main ingredients, was approved by the FDA in October 2010 as an orphan drug for the treatment of pseudobulbar affect (PBA)[3]. Quinidine is mainly used for anti-arrhythmia, and the clinical dose is 200-300mg, which is a reversible inhibitor of CYP2D6. However, reports have pointed out that AVP-923[5] has cardiotoxicity due to the addition of quinidine. AVP-786[6] is a compound drug with a halved dose of quinidine and deuterated dextromethorphan, which can block metabolic sites and is conducive to maintaining the exposure of dextromethorphan on the basis of reduced use of quinidine. Currently, AVP-786 is undergoing clinical trials for anxiety (Phase 3) and schizophrenia (Phase 2/3). In Phase 2 clinical studies, AVP-786 also includes indications for brain injury, impulse control disorders, severe depression, neurodegenerative diseases, etc.
In summary, dextromethorphan/bupropion is a fast-acting oral NMDA receptor antagonist with a variety of activities. The drug achieves pharmacological synergy by simultaneously targeting monoamines, NMDA receptors and sigma-1 receptors, reducing depression scores faster and more effectively than using bupropion alone. Dextromethorphan / bupropion has pharmacological characteristics similar to ketamine, making it a very promising new drug for the treatment of depression.

References
1.Stahl SM. Stahl’s Essential Psychopharmacology: Neuroscientific Basis and Practical Applications. 4th ed. New York, NY: Cambridge University Press; 2013.
2.Stephen M. Stahl et al; Dextromethorphan/Bupropion: A Novel Oral NMDA (N-methyl-d-aspartate) Receptor Antagonist withMultimodal Activity, CNS Spectrums (2019), 24, 461–466.
3. L. Nguyen et al. / Pharmacology & Therapeutics 159 (2016) 1-22.
4.Xue-mei Qin etal; Research on the Pathological Mechanism and Drug Treatment Mechanism of Depression; Current Neuropharmacology, 2015, 13, 514-523.
5.Dextromethorphan/Quinidine AVP 923, Dextromethorphan/Cytochrome P450-2D6 Inhibitor, Quinidine/Dextromethorphan. Drugs R D 2005; 6 (3): 174-177 ADIS R&D PROFILE 1174-5886/05/0003-0174/$34.95/0.
6.Rita Khoury et al; AVP-786 as a promising treatment option for Alzheimer’s Disease including agitation.https://doi-org.libproxy1.nus.edu.sg/10.1080/14656566.2021.1882995.