Compilation of 13 FDA-Approved ADC (Antibody-Drug Conjugate) Medications
In 2022, global sales of ADC (Antibody-Drug Conjugates) drugs exceeded 7 billion US dollars.
In 2023, this figure broke through the 10 billion mark, reaching 10.2 billion US dollars!
Compared to chemotherapy drugs, ADCs have advantages such as better tolerability, accurate target identification, and fewer side effects on non-cancerous cells, which have made them increasingly significant in the therapeutic field. At present, 13 types of ADCs have been approved by the United States Food and Drug Administration (FDA), with over 200 additional ADCs at various stages of clinical trials. This article will focus on introducing the 13 ADCs that have been approved for marketing by the FDA.
01
Gemtuzumab ozogamicin(GO)
Gemtuzumab ozogamicin (GO), developed by Pfizer, holds the distinction of being the first antibody-drug conjugate (ADC) to gain global market approval. This therapeutic agent is indicated for the treatment of acute myeloid leukemia (AML).
The structure of GO is comprised of a humanized monoclonal antibody that targets the CD33 antigen, conjugated to a potent cytotoxic agent, N-acetyl-γ-calicheamicin. This linkage is facilitated through a cleavable hydrazone bond, allowing for the targeted delivery of the cytotoxic payload.
The mechanism of action involves the binding of GO to CD33 antigens present on the surface of AML cells, leading to the formation of GO-CD33 complexes. These complexes are subsequently internalized by the AML cells, facilitating the release of the cytotoxic drug and inducing cell death.
Following promising results from three early-stage clinical trials, GO received approval from the U.S. Food and Drug Administration (FDA) in 2000, particularly for the treatment of CD33-positive AML patients over the age of 60.

However, subsequent safety concerns emerged from the Southwest Oncology Group (SWOG) S0106 study, which evaluated the efficacy of GO in adult AML patients under the age of 60. The study revealed a higher rate of treatment-related mortality in the GO group compared to the control group (5.5% versus 1.4%). Additional adverse effects associated with GO treatment included myelosuppression, infusion reactions, infections, bleeding, and hepatotoxicity. As a result, Pfizer voluntarily withdrew the product from the market in June 2010.
Subsequent data indicated that GO demonstrated a favorable improvement in prognosis for elderly AML patients not suitable for intensive chemotherapy, with the toxicity profile being manageable. Based on these findings, the FDA reapproved GO in 2017.
Further research has shown that when combined with fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin, GO maintains its efficacy and safety profile, particularly in the initial treatment of CD33-positive AML patients.
02
Brentuximab vedotin (BV)
Brentuximab vedotin (BV), developed by Seagen Inc. and co-developed with Takeda Pharmaceutical Company, represents the second antibody-drug conjugate (ADC) to have received approval for medical use.
BV is employed in the treatment of Hodgkin's lymphoma (HL) and anaplastic large cell lymphoma (ALCL). Its structure comprises three integral components: an anti-CD30 chimeric monoclonal antibody (brentuximab), a protease-cleavable dipeptide linker (mc-VC-PABC), and the microtubule-disrupting agent monomethyl auristatin E (MMAE).
The mechanism of action for Brentuximab vedotin uniquely targets the CD30 antigen, which is expressed on the surface of the affected cells. By doing so, BV delivers a potent cytotoxin directly to the malignant cells, sparing most normal tissues.
Commercially, BV achieved significant success, with sales in the United States and Canada reaching $628 million in 2019, and ¥52.7 billion in other markets. In 2022, sales surged to $1.469 billion and by 2023, an increase to $1.65 billion was recorded. Projections suggest that global sales of BV could reach $1.8 billion by the year 2026.
BV's efficacy and safety profile were evaluated in Phase I studies for the treatment of HL and ALCL. The majority of adverse events were classified as Grade 1 or 2, with common side effects including fatigue, fever, diarrhea, nausea, and neutropenia. In a Phase II clinical trial, BV demonstrated effectiveness in 75% and 87% of patients with HL (102 patients total) and ALCL (30 patients total), respectively. Furthermore, BV has been noted to alleviate symptoms such as itching and pain caused by lymphomas without negatively impacting patients' quality of life.
In 2018, the U.S. Food and Drug Administration (FDA) approved the use of BV in combination with CHP (cyclophosphamide, doxorubicin, and prednisone) for the treatment of adult patients with previously untreated systemic ALCL or other CD30-expressing cancers.
03
Trastuzumab emtansine (T-DM1)
Trastuzumab emtansine(T-DM1), is an innovative targeted therapy developed through a collaboration between Genentech and ImmunoGen. Structurally, T-DM1 is a complex molecule that links the monoclonal antibody trastuzumab to the cytotoxic agent emtansine (DM1) via a thioether linker. This design facilitates the targeted delivery of a potent cytotoxic molecule directly to HER2-positive cancer cells.
Indicated for the treatment of HER2-positive cancers, T-DM1 works by selectively binding to HER2 overexpressing tumor cells. Its mechanism of action involves the internalization of the trastuzumab-DM1 complex and subsequent release of the DM1 molecule, which possesses high anti-tumoral activity. This approach significantly reduces toxic side effects and enhances the targeting of the therapy, leading to a more precise attack on cancer cells while sparing healthy tissues.
In terms of its market performance, T-DM1 achieved global sales of $2.136 billion in 2022 and saw a 4% growth in 2023, reaching $2.222 billion in sales. Although its use is confined to treating HER2-positive breast cancer and it faces competition from biosimilars, T-DM1 is projected to maintain a steady market presence with forecasted sales of $2.3 billion by 2026.
Clinical confirmation of T-DM1's efficacy was provided by the EMILIA study organized by Verma and colleagues. This research demonstrated the enhanced therapeutic effectiveness of T-DM1 in patients with HER2-positive advanced or metastatic breast cancer when compared to treatments using trastuzumab and docetaxel, leading to improved patient safety and fewer adverse reactions. Side effects were generally mild, with fatigue and nausea each reported in 49.3% of patients, elevated AST in 43.5%, fever in 40.6%, and headaches in 40.6% of cases.
T-DM1's success in the clinical setting underpins its status as an important therapeutic option for patients with HER2-positive breast cancer, offering a paradigm of how targeted therapies can vastly improve both the efficacy and quality of cancer treatment.
04
Inotuzumab ozogamicin (InO)
Inotuzumab Ozogamicin (InO) is a targeted cancer therapy developed by Wyeth Pharmaceuticals Inc. This therapeutic agent is a conjugate formed by linking a human IgG4 monoclonal antibody that targets CD22, with the cytotoxic chemotherapeutic agent calicheamicin. The conjugation is facilitated via acid-mediated labile splicing, allowing the drug to deliver its cytotoxic payload directly to CD22-expressing cells.
InO is specifically indicated for the treatment of acute lymphoblastic leukemia (ALL), a type of cancer in which the bone marrow makes too many lymphocytes, a subtype of white blood cells. The mechanism of InO involves binding to tumor cells that express the CD22 antigen, thereby initiating the internalization of the InO-CD22 complex. This process results in the hydrolytic cleavage between the subunits of the complex. The activated cytotoxic drug then induces double-stranded DNA breaks, culminating in cell cycle arrest and subsequent cell death. CD22 is a 135-kDa transmembrane sialoglycoprotein that is nearly ubiquitously present in the cytoplasm of all B-cells and is specifically expressed on B-cells, primarily on those that co-express immunoglobulin M (IgM) and immunoglobulin D (IgD).
InO has proven to be an effective therapeutic option for patients with relapsed or refractory ALL, offering more patients the opportunity to undergo stem cell transplantation and potentially achieve long-term survival. However, the treatment is not without side effects, with the most common adverse reactions including neutropenia in 28% of patients, increased AST levels in 26%, nausea in 21%, vomiting in 17%, fatigue in 15%, and febrile neutropenia also in 15%. Despite these side effects, InO remains a critical asset in the management of advanced ALL, providing hope for patients facing this challenging disease.
05
Moxetumomab pasudotox (MP)
Moxetumomab Pasudotox (MP), developed by AstraZeneca, represents the first medicinal therapy approved for the treatment of hairy cell leukemia (HCL). It is a unique fusion protein consisting of three components: a monoclonal antibody targeting CD22, a 38 kDa fragment of Pseudomonas exotoxin A, and a linker mc-VC-PABC. MP is specifically indicated for adult patients with relapsed or refractory hairy cell leukemia who have not responded to at least two prior systemic therapies, including purine nucleoside analogs.
The most common high-grade adverse reactions associated with MP include lymphocyte count decrease (20%), asymptomatic hypophosphatemia (10%), and anemia (10%). Updated clinical data has confirmed that Moxetumomab Pasudotox demonstrates high rates of durable response and minimal residual disease in heavily pretreated HCL patients. Due to its proven efficacy and manageable safety profile, it is considered a safe and viable treatment option for this patient population.
06
Polatuzumab vedotin (PV)
Polatuzumab vedotin (PV), developed by Genentech, is an innovative therapeutic agent that functions by connecting an antibody targeting CD79b to the cytotoxic agent monomethyl auristatin E (MMAE) through a cleavable dipeptide linker (mc-VC-PABC). This structure enables PV to deliver targeted chemotherapy to B cells, which are implicated in certain cancers.
Indicated for adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), Polatuzumab vedotin operates by inducing degradation of the MCL-1 protein, a member of the BCL-2 family, through the ubiquitin/proteasome system. This action contributes to the treatment's capacity to promote cancer cell death.
In terms of performance, Polatuzumab vedotin (marketed as Polivy) has shown impressive growth, with sales surging by 108% in 2023 to reach a revenue of $946 million. The global sales for PV are anticipated to hit $850 million by the year 2026.
The efficacy of Polatuzumab vedotin is further enhanced when used in combination with venetoclax and anti-CD20 antibodies like obinutuzumab or rituximab. This particular combination capitalizes on the MCL-1 antagonistic action to induce remission in non-Hodgkin’s lymphoma (NHL). Notably, the remission has been observed to persist even after cessation of therapy, with a significant proportion of patients responding positively to the treatment.
Despite these promising results, a separate study has indicated that a PV combination with rituximab and lenalidomide failed to meet the pre-established efficacy threshold in patients with relapsed/refractory DLBCL. Common severe adverse reactions (Grade 3-4) experienced by patients included neutropenia and thrombocytopenia, highlighting the need to balance efficacy with the management of toxicity.
07
Enfortumab vedotin (EV)
Enfortumab Vedotin (EV) is a therapeutic agent developed by Astellas Pharma and Pfizer, designed to target and treat locally advanced or metastatic urothelial cancer (la/ mUC). The drug's composition integrates an antibody specific to Nectin-4 with the cytotoxic agent monomethyl auristatin E (MMAE), linked via a cleavable connector. This strategic assembly allows EV to deliver its cancer-killing payload directly to the cancer cells.
Upon administration, EV binds to Nectin-4, a protein frequently overexpressed on urothelial cancer cells, forming a complex. This complex is then endocytosed by cells expressing Nectin-4, which allows the subsequent release of MMAE inside the cell. Once released, MMAE binds to tubulin within the cellular microtubules, leading to the disruption of the microtubule network. This results in cell cycle arrest and prompts apoptosis, effectively eradicating the cancer cells.
The financial performance of EV has been remarkable, with global sales reaching $754 million in 2022 and projections suggesting an increase to $1.03 billion in 2023. Expectations for the year 2026 forecast global revenues potentially climbing to $3.5 billion.
In the phase I dose-escalation study EV-101, involving 112 mUC patients who received monotherapy with EV, the outcomes were promising. The study reported an objective response rate of 43%, as assessed by researchers, with a median duration of response lasting 7.4 months. The median survival rate stood at 12.3 months, with a one-year survival rate of 51.8%. However, treatment-related adverse events (TRAEs) were observed, occurring in 30% of the patients. The most common TRAEs included fatigue, hair loss, decreased appetite, taste disturbances, nausea, peripheral sensory neuropathy, pruritus, and diarrhea.
Furthermore, the U.S. Food and Drug Administration (FDA) recognized the combination therapy of EV with pembrolizumab (EV+P) as a breakthrough therapy. It also authorized EV as a first-line treatment for la/ mUC patients who are ineligible for cisplatin therapy, marking a significant milestone in the management of this challenging cancer.
08
Trastuzumab deruxtecan (T-DXd)
Trastuzumab deruxtecan, also known as T-DXd, is an innovative therapeutic agent developed by Daiichi Sankyo Company. This conjugate drug combines trastuzumab—a humanized monoclonal antibody that targets HER2—with a derivative of exatecan, which acts as a topoisomerase I inhibitor. The result is a powerful targeted treatment designed for patients with unresectable or metastatic HER2-positive breast cancer.
The mechanism of action for T-DXd sets it apart from its predecessor, T-DM1, by enabling the delivery of a greater payload of cytotoxic medication directly to the cancer cells. Moreover, T-DXd's superior membrane permeability allows for the "bystander effect," wherein it can effectively kill adjacent tumor cells not directly targeted by the antibody, providing an additional therapeutic benefit.
T-DXd has shown remarkable promise and is projected to become the highest-grossing antibody-drug conjugate (ADC) with estimated sales of $6.2 billion by the year 2026. Its versatility across multiple subgroups of breast cancer, including HER2-positive, HR+/HER2-negative, and triple-negative types, combined with its long treatment duration, underline its significant potential in the oncology market.
In a phase I clinical trial that was open-label, employed dose escalation, and expanded the dosing cohort, researchers assessed T-DXd’s safety, tolerability, and anticancer activity in patients with advanced solid tumors expressing HER2. Findings from this study indicated that out of 111 patients treated, 66 experienced a pharmacologic response, and disease control was achieved in 104 of the patients. However, all participants experienced at least one adverse event. Severe adverse reactions occurring at grade 3 or above included anemia (17% of patients), neutropenia (14%), leukopenia (9%), and thrombocytopenia (8%). Furthermore, serious adverse reactions were reported by 19% of the patients, with 20 individuals developing interstitial lung disease, pneumonitis, or pneumonia.
09
Sacituzumab govitecan (SG)
Sacituzumab govitecan (SG), developed by Immunomedics, is the only globally approved Trop-2 targeting antibody-drug conjugate (ADC) medication available on the market. This innovative drug is comprised of a trio of components: an anti-Trop-2 antibody, an SN-38 drug payload (which is an active metabolite of the chemotherapy drug irinotecan), and a CL2A linker that chemically binds the two.
SG is indicated for the treatment of adult patients with metastatic triple-negative breast cancer (TNBC), a type of cancer that is notoriously challenging to manage due to the absence of estrogen and progesterone receptors and HER2 protein. The mechanism of action of SG is centered around its specificity for Trop-2, short for trophoblast cell-surface antigen-2—a 40-kDa glycoprotein, also referred to as tumor-associated calcium signal transducer-2—which plays a crucial role in the development and metastasis of various solid tumors. By targeting Trop-2 with its toxic payload of SN-38, SG distinguishes itself by reducing off-target toxicities due to its high drug-to-antibody ratio (7-8:1).
Commercially known as Trodelvy, Sacituzumab govitecan has demonstrated impressive financial performance, with sales in its first full year on the market reaching $380 million. In 2022, worldwide sales climbed to $681 million, and in 2023, Trodelvy crossed the billion-dollar threshold for the first time, registering a 56% year-on-year increase to $1.063 billion. The drug’s sales are projected to continue to grow, with an anticipated $1.1 billion in revenue by 2026.
From a clinical standpoint, the drug's efficacy and safety in treating locally advanced or metastatic urothelial cancer patients who have previously failed platinum-based and immune checkpoint inhibitor (ICI) therapies have been substantiated by clinical trial NCT03547973. Moreover, studies have suggested that cells resistant to EV (a type of chemotherapy) are still sensitive to Sacituzumab govitecan, implying its potential efficacy across most bladder cancer subtypes, including those treated with EV. This represents a significant advancement in treatment options for patients with urothelial cancer.
10
Belantamab mafodotin (Blenrep)
Belantamab mafodotin(Blenrep), is an innovative cancer treatment developed by GlaxoSmithKline (GSK). It has gained recognition as the world's first approved therapy that targets B-cell maturation antigen (BCMA). This pioneering therapy is an antibody-drug conjugate (ADC) that consists of an IgG1 antibody linked to the microtubule-disrupting agent monomethyl auristatin F (MMAF) via a maleimidocaproyl (mc) linker that is susceptible to protease cleavage.
Blenrep is indicated for use as a monotherapy in adult patients with relapsed or refractory multiple myeloma (R/R MM) who have previously undergone at least four different therapy regimens. It is specifically designed for patients whose disease is resistant to at least one proteasome inhibitor, one immunomodulatory agent, and one CD38 monoclonal antibody, and for those who have seen their disease progress after the most recent treatment.
Despite its groundbreaking potential, Blenrep faced a significant setback in November 2022 when a confirmatory Phase III trial failed to meet expectations, leading the FDA to request GSK to withdraw the drug from the US market within the same month. Furthermore, while Blenrep achieved sales of $122 million in 2021 and saw an increase to $143 million in 2022, the treatment's market outlook remains challenging due to competition from several other BCMA-targeting products.
Nevertheless, a silver lining appeared on November 27, 2023, when Blenrep's use as a second-line therapy for R/R MM achieved a significant milestone in the Phase III DREAMM-7 clinical trial. The trial met its primary endpoint of progression-free survival (PFS) at an interim analysis, providing a glimmer of hope for its future market prospects amidst a highly competitive landscape.
11
Loncastuximab tesirine (LT)
Loncastuximab Tesirine (LT), developed by ADC Therapeutics, is an innovative therapeutic agent designed for the treatment of relapsed or refractory large B-cell lymphoma (LBCL). It is structured as a humanized anti-human CD19 monoclonal antibody linked through a valine-alanine cleavable linker to a pyrrolobenzodiazepine (PBD) dimer toxin. This unique composition allows LT to specifically target and deliver cytotoxic agents to CD19-expressing cells.
LT is indicated for patients who require second-line or later systemic therapy for relapsed or refractory LBCL, including not otherwise specified diffuse large B-cell lymphoma (DLBCL), DLBCL arising from low-grade lymphoma, and high-grade B-cell lymphoma.
The most commonly observed adverse reactions associated with the treatment include neutropenia (a decrease in the number of neutrophils), thrombocytopenia (a decrease in platelets), and elevated levels of gamma-glutamyltransferase (an enzyme involved in amino acid transfer). Reports show that 39% of patients experienced severe adverse events, which included severe neutropenia, pleural effusion, anemia, pericardial effusion, and non-cardiac chest pain. As with any potent medical therapy, careful monitoring and management of these adverse reactions are necessary to optimize patient outcomes.
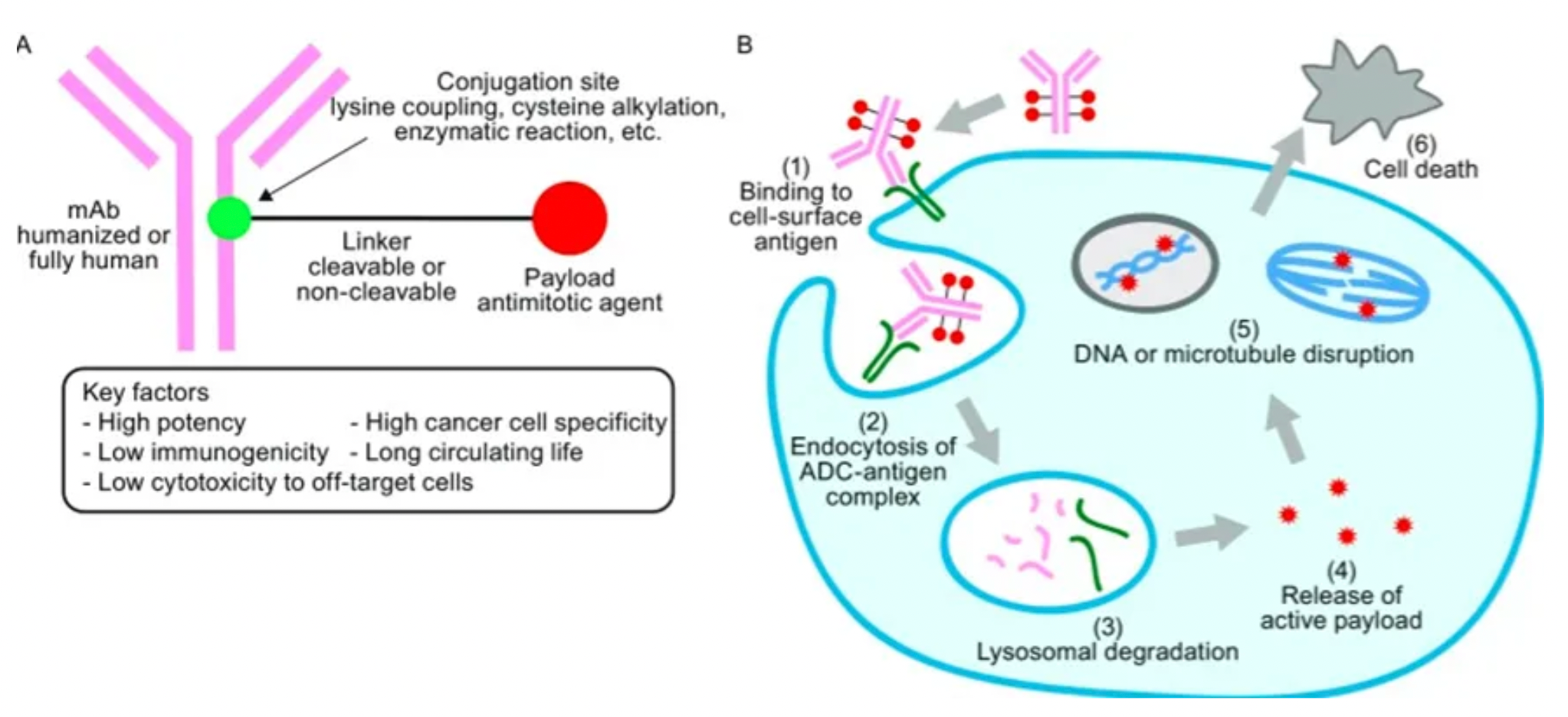
12
Tisotumab vedotin (TV)
Tisotumab Vedotin (TV) is a cutting-edge anticancer drug developed by Seagen, noteworthy as the only approved antibody-drug conjugate (ADC) for the treatment of cervical cancer. This innovative therapeutic is comprised of a fully human monoclonal antibody that specifically targets tissue factor (TF), which is conjugated to the cytotoxic agent monomethyl auristatin E (MMAE).
TF, also known as coagulation factor III, thromboplastin, or CD142, is a transmembrane glycoprotein that plays a vital role in initiating the extrinsic coagulation cascade. Its overexpression on the surface of cancer cells is implicated in tumor growth, angiogenesis, and metastasis. The monoclonal antibody component of TV specifically binds to TF, leading to the internalization of MMAE and the subsequent induction of cytotoxic effects within the cancerous cells.
TV is indicated for the treatment of recurrent or metastatic cervical cancer (r/mCC) in adult patients who have experienced tumor progression following chemotherapy. The safety profile, tolerability, pharmacokinetics, and antitumor activity of TV have been evaluated in an open-label, phase I/II study known as innovaTV 201 (NCT02001623), which focused on patients with TF-expressing metastatic solid tumors. The most common adverse reactions observed during this study included alopecia, epistaxis, nausea, conjunctivitis, fatigue, and dry eye syndrome.
13
Mirvetuximab soravtansin (MIRV)
Mirvetuximab soravtansin (MIRV) is an experimental cancer treatment being developed by ImmunoGen. It is composed of a folate receptor alpha (FRα)-targeting monoclonal antibody that is chemically linked to a microtubule inhibitor through a cleavable linker.
MIRV is designed to treat adult patients with FRα-positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer who have previously undergone one to three systemic treatment regimens. Folate receptor alpha (FRα) has a high affinity for folate and facilitates its transport into the cytoplasm via endocytosis. Although FRα is typically expressed at low levels in normal tissues, it is frequently overexpressed in a variety of epithelial tumors, including epithelial ovarian cancer, endometrial adenocarcinoma, triple-negative breast cancer (TNBC), and non-small cell lung cancer (NSCLC). Through its monoclonal antibody, MIRV specifically targets FRα, delivering the potent microtubule inhibitor directly to cancer cells that express this receptor.
Current studies are focused on the safety and efficacy of MIRV in combination with other drugs for the treatment of patients with ovarian cancer. This includes evaluating the safety and anti-tumor activity of MIRV in combination with carboplatin for patients with recurrent, platinum-sensitive epithelial ovarian or fallopian tube cancer. Common adverse reactions observed with MIRV include diarrhea, blurred vision, nausea, and fatigue.
How to search for and analyze the development progress of ADC pharmaceuticals?
If you want to learn about the latest developments in ADC drugs, you can use the drug search module of the Synapse database. This module supports searching for ADC drugs by classification through Targeting Moiety, Linker, and Payload.
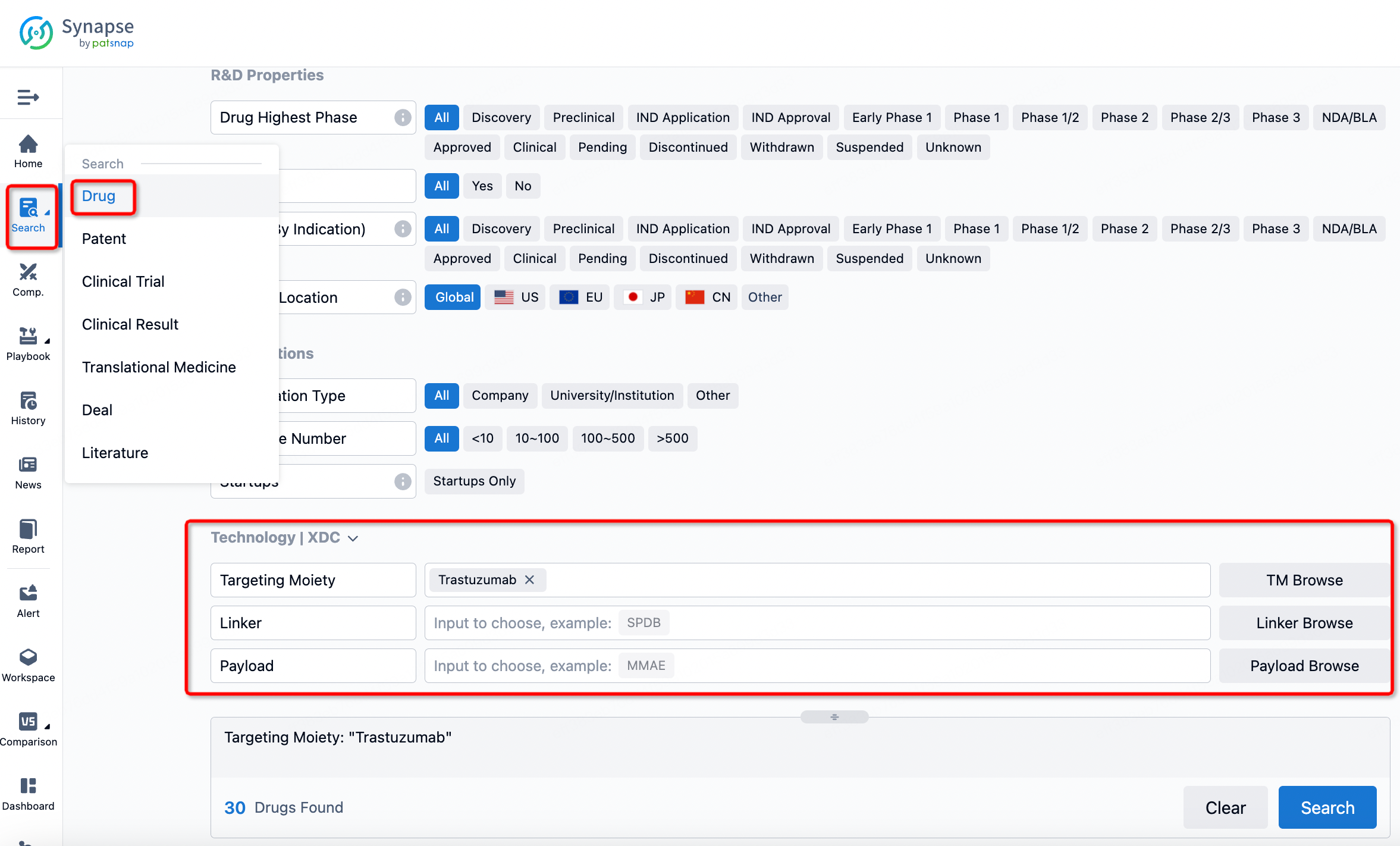
On the search results page, you can easily review information related to the ADC's technical category of the drugs.
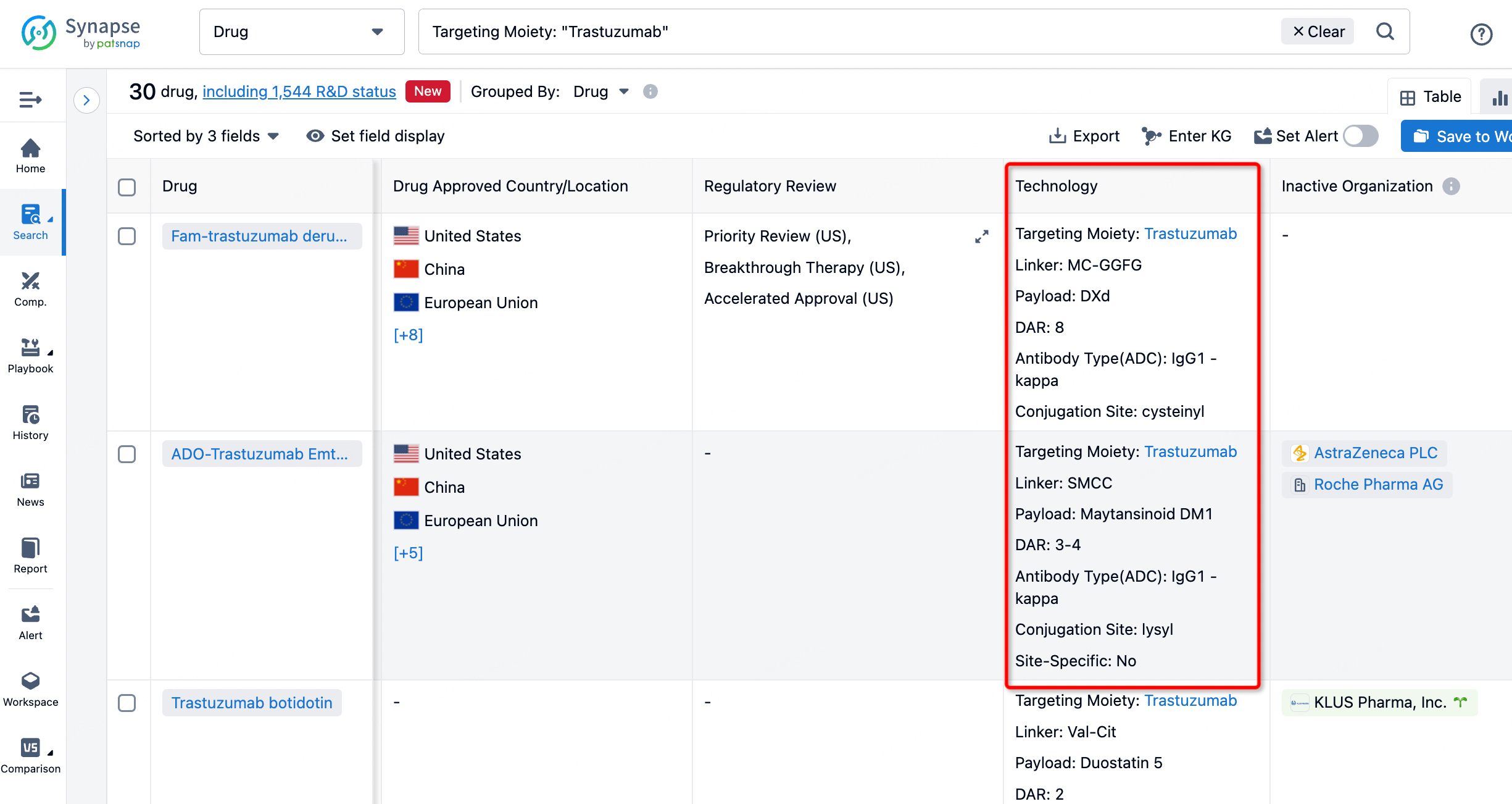
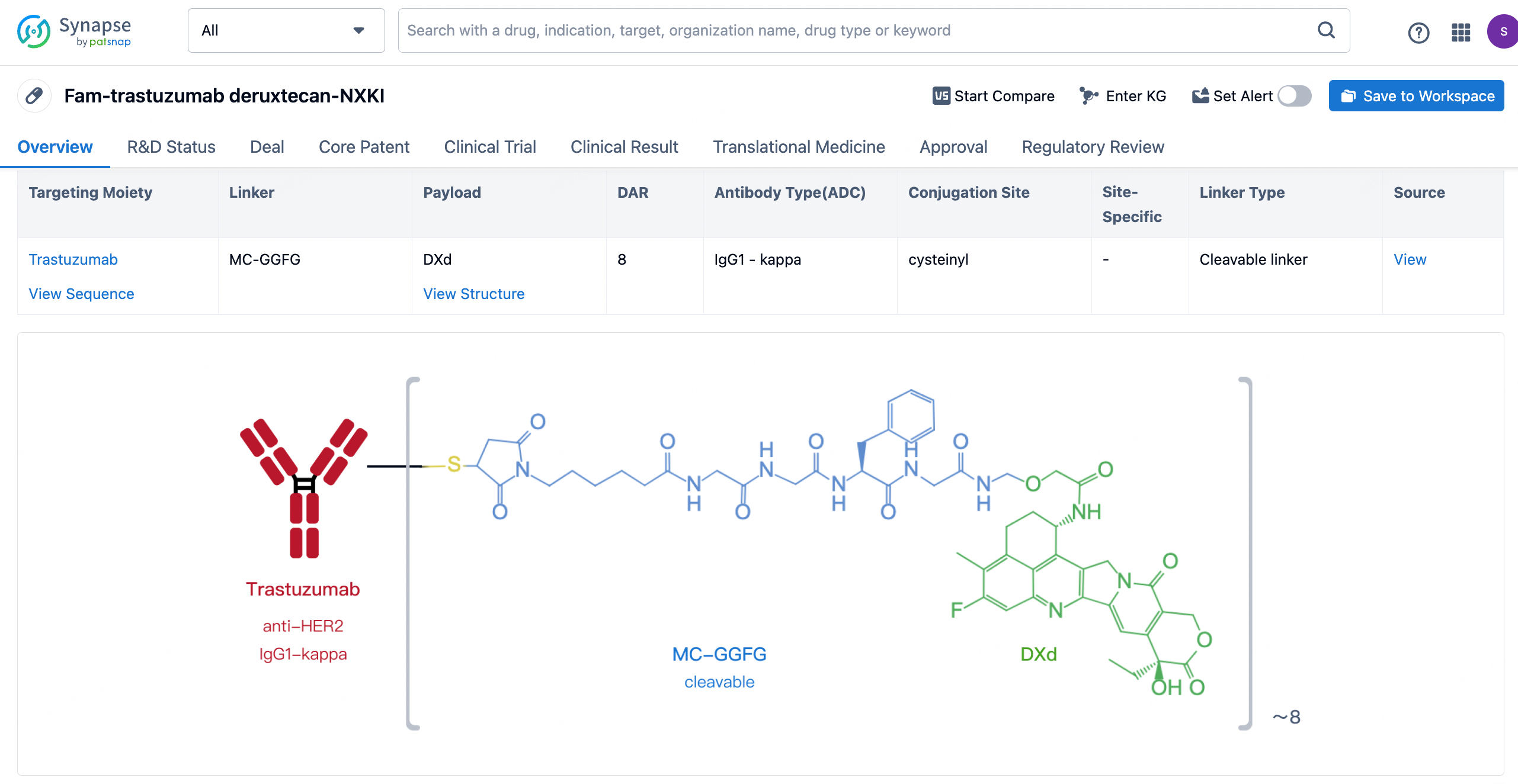
After clicking to enter the drug details page, you can also effortlessly obtain structural information about the ADC drug.
Click on the image below to embark on a brand new journey of drug discovery!
Reference
- 1. Liu K, Li M, Li Y, Li Y, Chen Z, Tang Y, Yang M, Deng G, Liu H. A review of the clinical efficacy of FDA-approved antibody‒drug conjugates in human cancers. Mol Cancer. 2024 Mar 23;23(1):62. doi: 10.1186/s12943-024-01963-7. PMID: 38519953;
- 2. Indini A, Rijavec E, Grossi F. Trastuzumab Deruxtecan: Changing the Destiny of HER2 Expressing Solid Tumors. Int J Mol Sci. 2021 Apr 30;22(9):4774. doi: 10.3390/ijms22094774. PMID: 33946310; PMCID: PMC8125530.
- 3. Fenwarth, L.; Fournier, E.; Cheok, M.; Boyer, T.; Gonzales, F.; Castaigne, S.; Boissel, N.; Lambert, J.; Dombret, H.; Preudhomme, C.; et al. Biomarkers of Gemtuzumab Ozogamicin Response for Acute Myeloid Leukemia Treatment. Int. J. Mol. Sci. 2020, 21, 5626.
- 4. Dhillon S. Moxetumomab Pasudotox: First Global Approval. Drugs. 2018 Nov;78(16):1763-1767. doi: 10.1007/s40265-018-1000-9. Erratum in: Drugs. 2019 Jan;79(1):105. PMID: 30357593;
- 5. Harbeck N, Gluz O, Christgen M, et al. De-escalation strategies in human epidermal growth factor receptor 2 (HER2)–positive early breast cancer (BC): final analysis of the west german study group adjuvant dynamic marker-adjusted personalized therapy trial optimizing risk assessment and therapy response prediction in early BC HER2- and hormone receptor–positive phase II randomized trial—efficacy, safety, and predictive markers for 12 weeks of neoadjuvant trastuzumab emtansine with or without endocrine therapy (ET) versus trastuzumab plus ET. J Clin Oncol. 2017 Jul 6. doi: 10.1200/JCO.2016.71.9815
- 6. Tsuchikama K, An Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell. 2018 Jan;9(1):33-46. doi: 10.1007/s13238-016-0323-0. Epub 2016 Oct 14. PMID: 27743348; PMCID: PMC5777969.



