Idiopathic Pulmonary Fibrosis: Current Clinical Research Status Targeting GPCR
In September last year, the South Korean biopharmaceutical company GPCR Therapeutics published a research paper in "Cell Communication and Signaling" about the direct interaction between two different GPCRs (CXCR4 and LPA1). Through various in vitro experiments, the company discovered that the efficacy of LPA1 inhibitors could be enhanced to its maximum potential by inhibiting CXCR4, thereby providing scientific evidence that the combination of CXCR4-LPA1 inhibitors might be a necessary condition for comprehensive antifibrotic activity. Based on this discovery, GPCR Therapeutics filed a provisional patent application for a combined therapeutic method involving CXCR4-LPA1 inhibitors.

GPCR Therapeutics will collaborate with Bridge Biotherapeutics to jointly develop and commercialize this combined therapy. Bridge Biotherapeutics is conducting a phase 2a multinational clinical trial for the autonomic sympatho-inhibitor BBT-877, intended for the treatment of idiopathic pulmonary fibrosis (IPF), and is accelerating the development of its proprietary IPF product pipeline.
About Idiopathic Pulmonary Fibrosis
Idiopathic Pulmonary Fibrosis (IPF) is a progressive chronic lung disease that leads to scarring (fibrosis) of the lungs, resulting in stiffening and loss of elasticity of the lung tissues, making breathing increasingly difficult. The efficiency of oxygen transfer in the lungs decreases, thus the process of oxygen passing from the alveoli to the blood during inhalation is compromised in IPF patients. The lung damage caused by IPF is irreversible, and currently, there are no treatment methods available that can stop or reverse the formation of scarring.
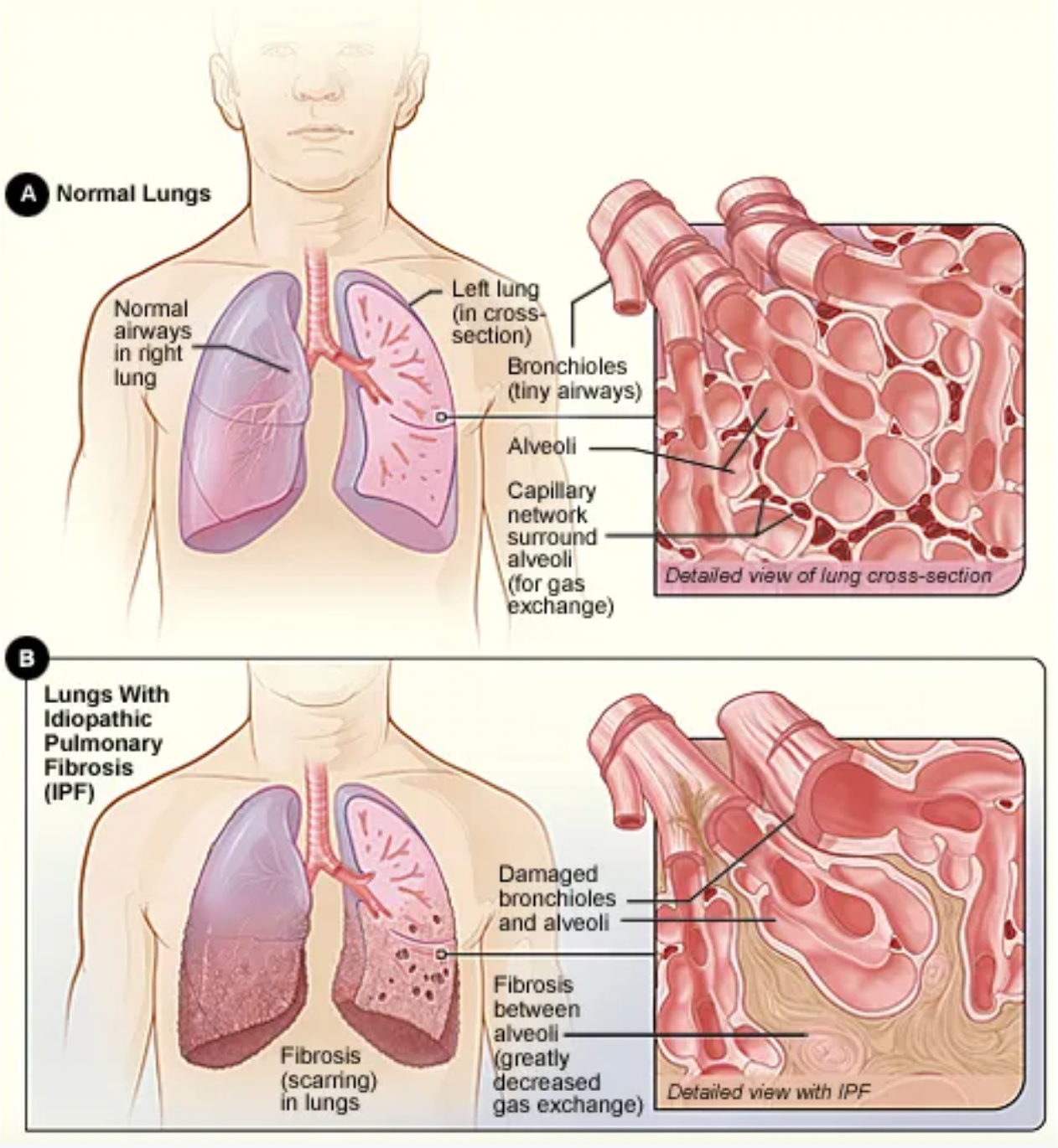
The prognosis of Idiopathic Pulmonary Fibrosis is generally poor and the progression of the disease is difficult to predict. For some patients, the disease may progress slowly, whereas for others, it may advance more rapidly. On average, the life expectancy of patients is relatively short. Even in the United States, which has some of the world's leading medical conditions, there are approximately 495 cases per 100,000 people, with a median survival estimated at 2-5 years post-diagnosis.
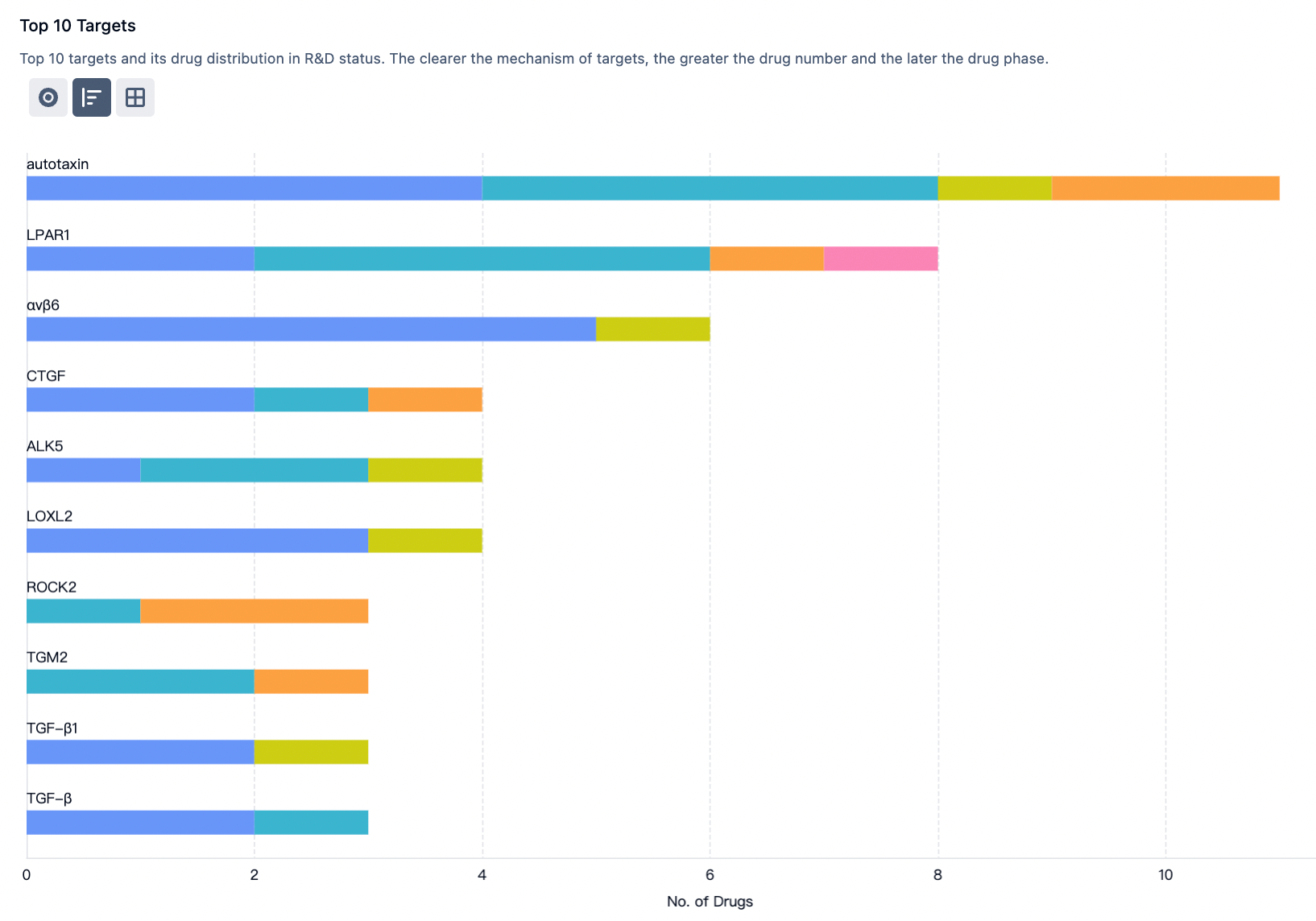
According to statistics from the Synapse database, there are a total of 341 R&D pipelines globally in the direction of idiopathic pulmonary fibrosis, with 4 approved drugs on the market, 6 projects in phase III clinical trials, 40 projects in phase II clinical trials, and 45 projects in phase I clinical trials.
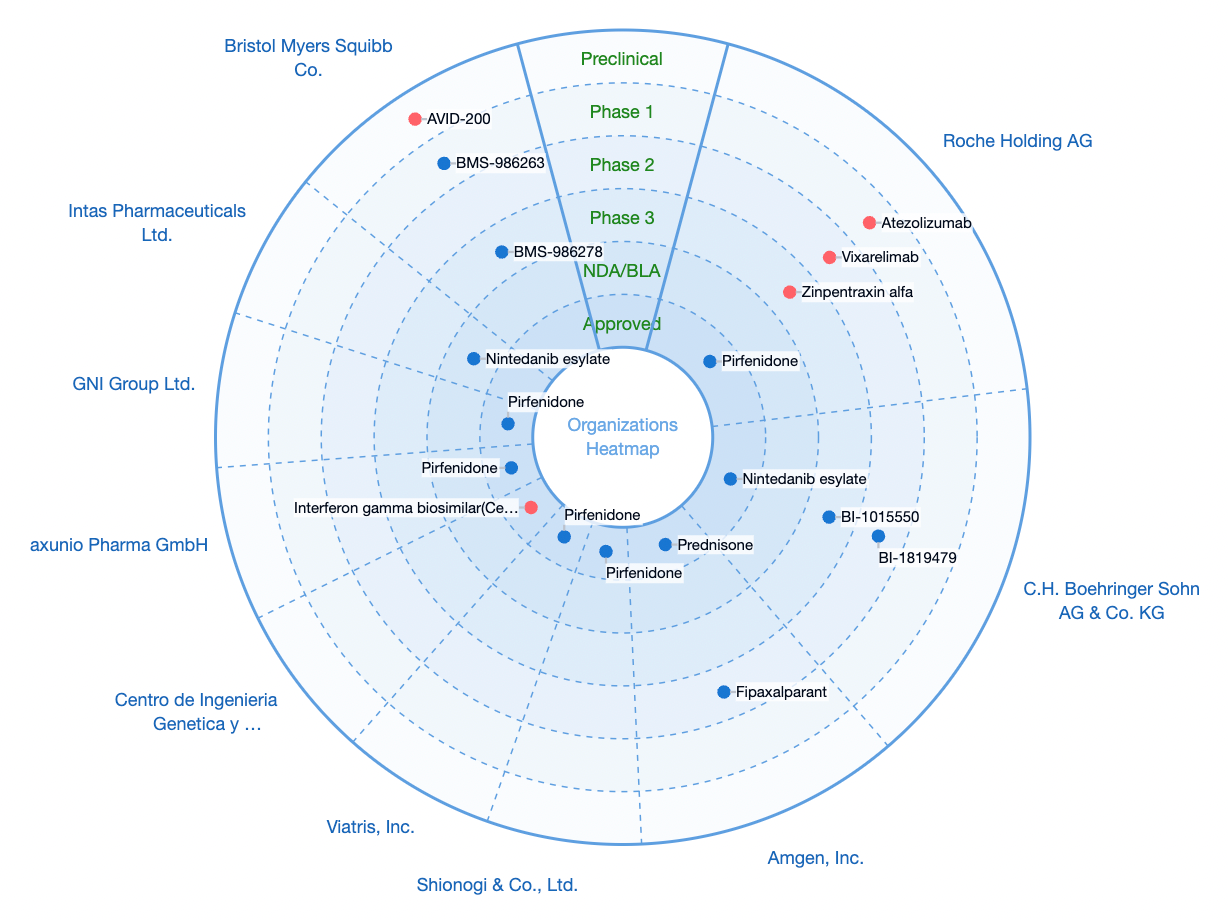
GPCR-class targets that are popular research targets for this clinical indication include autotaxin, LPAR1, αvβ6, etc.
BMS-986278 is a lysophosphatidic acid receptor 1 (LPAR1) antagonist developed originally by BMS Company. Extensive research indicates that idiopathic pulmonary fibrosis (IPF) can be mediated by lysophosphatidic acid (LPA), which acts through several of the six known lysophosphatidic acid receptor-mediated downstream signaling pathways.

The first generation LPAR1 antagonist developed by BMS Company, represented by BMS-986020, exhibited hepatobiliary toxicity and increased plasma bile acids in a phase II clinical trial. Subsequently, BMS Company conducted preclinical studies on the mechanism of hepatic toxicity with BMS-986020 and two structurally different LPA1 antagonists (BMS-986234 and BMS-986278), further confirming that the second-generation LPA1 antagonist BMS-986278 did not show hepatobiliary toxicity in preclinical and clinical safety studies. BMS-986278 is currently in phase II clinical development for the treatment of IPF and progressive fibrotic interstitial lung diseases.
In addition to BMS, companies including Sanofi, UBE, Novo Nordisk, and DJS Antibodies have also been involved in the development of drugs targeting LPAR1 for the treatment of idiopathic pulmonary fibrosis.
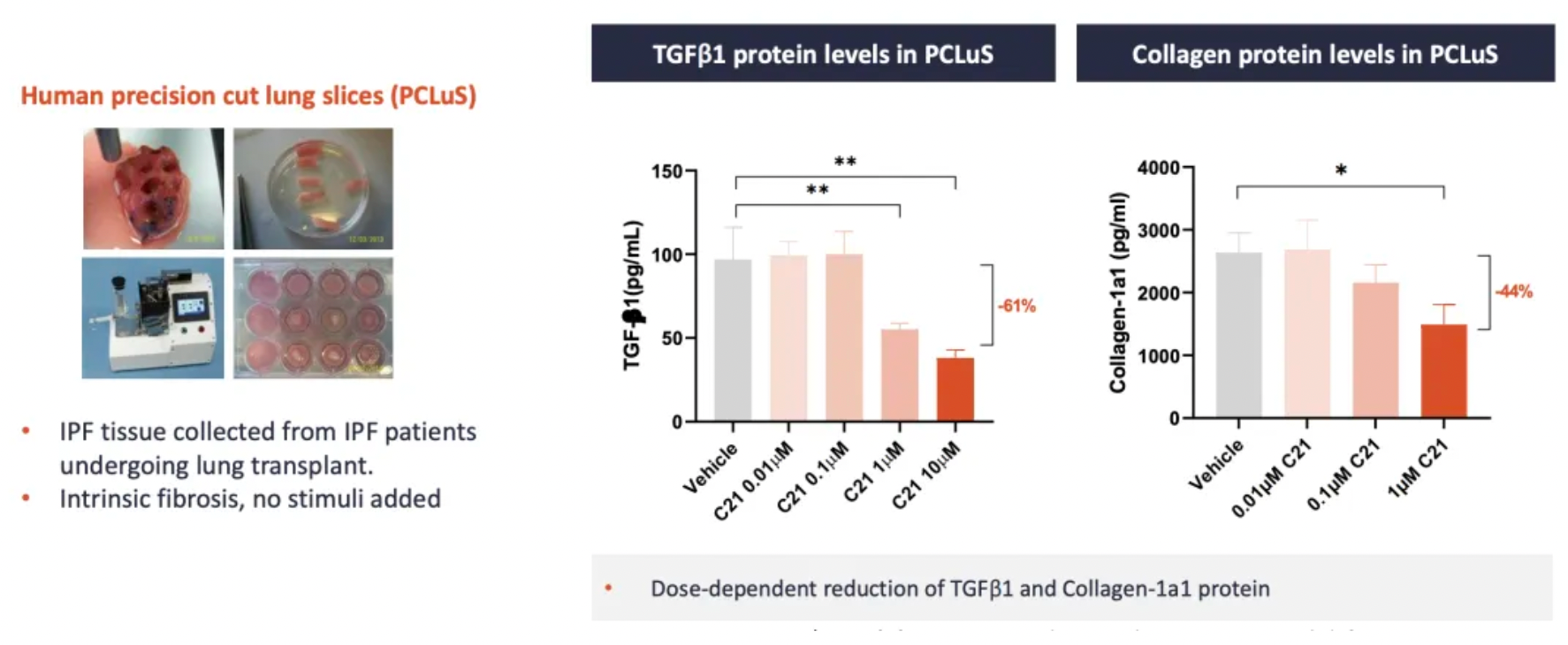
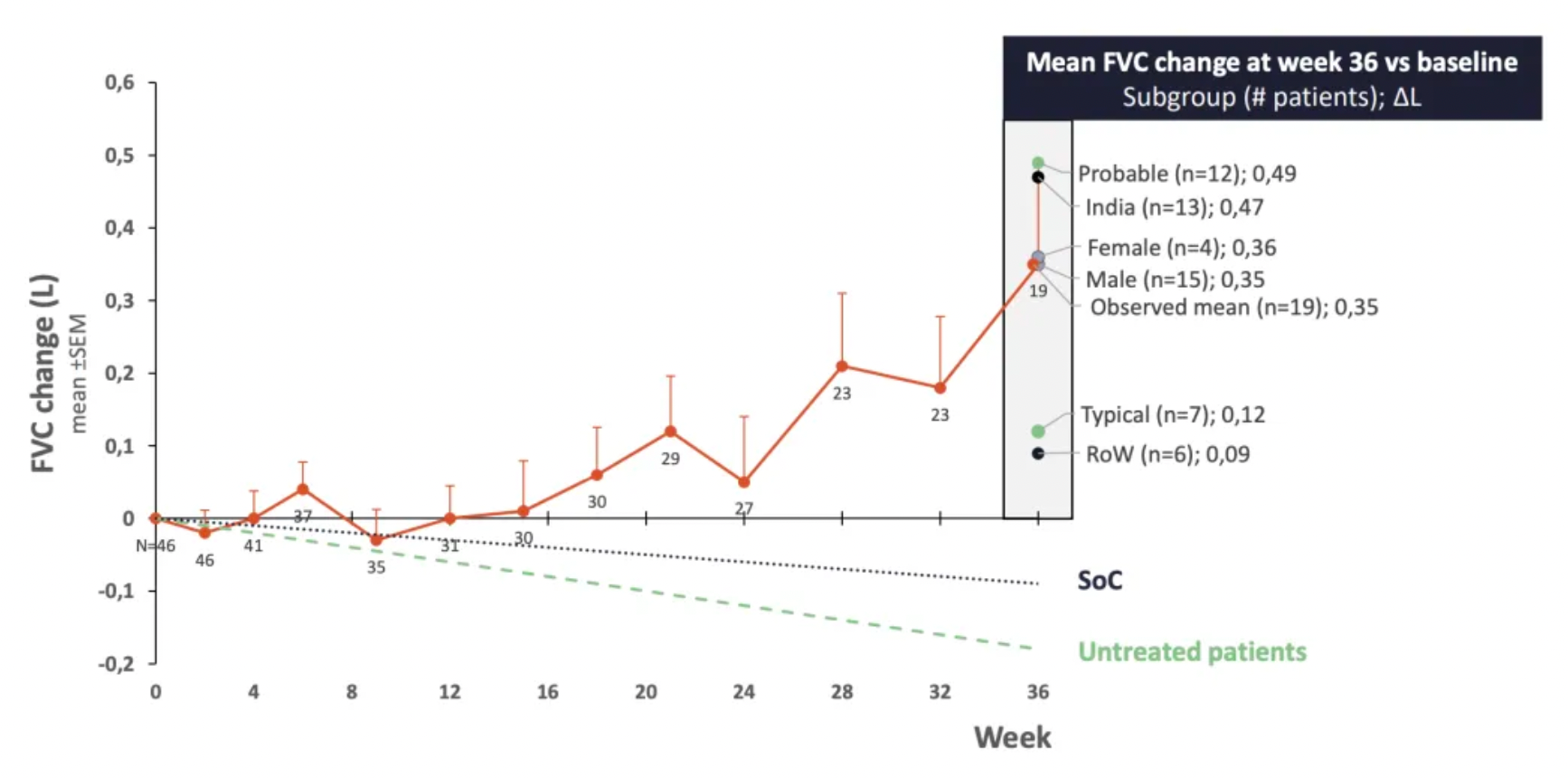
VP01 (C-21) is an AT2R agonist developed by Vicore Pharma. The company recently published research data demonstrating that C21 can reduce the content of TGFβ1 and collagen in human IPF lung slices.
In a phase 2a clinical trial, C21 showed good safety and efficacy in newly treated IPF patients, maintaining stable forced vital capacity over a 36-week treatment period.
Apart from Vicore Pharma, companies including MorphoSys and Emeriti Bio are also strategizing on the development of therapeutics targeting the AT2R for the treatment of idiopathic pulmonary fibrosis.
In 2017, a team from Genentech published a research paper in the journal Thorax, discussing CXCL14 as a potential biomarker for Hedgehog signaling in idiopathic pulmonary fibrosis.

Compared to the control group, there is a significant increase in the expression of Hedgehog-associated genes in the lung tissue of patients with Idiopathic Pulmonary Fibrosis (IPF). The most upregulated gene in IPF lung biopsy tissues and fibroblasts subjected to SHh stimulation in vitro was CXCL14, a gene that encodes a soluble secreted chemokine whose expression is inhibited in vitro by vismodegib. Overexpression of SHh in the lungs of mice induces the expression of CXCL14. Circulating levels of CXCL14 protein in the plasma of IPF patients are significantly higher than those in the control group. In cancer patients, circulating levels of CXCL14 decreased significantly after treatment with vismodegib.
Therefore, the IL-13 inhibitor Lebrikizumab, originally developed by Genentech, is considered to have potential for the treatment of IPF.
Treprostinil is a prostacyclin receptor agonist originally developed by United Therapeutics for the treatment of pulmonary arterial hypertension to alleviate exercise-related symptoms. Treprostinil is a stable, synthetic analog of prostacyclin that promotes vasodilation of the pulmonary and systemic arterial beds and inhibits platelet aggregation. It helps reduce symptoms in patients with Pulmonary Arterial Hypertension (PAH) and patients with pulmonary hypertension associated with interstitial lung disease. The FDA approved Treprostinil for the treatment of PAH in 2002.
The first drug approved for the treatment of PAH was epoprostenol, an artificially synthesized prostacyclin, which significantly improves the quality of life for patients. However, the use of epoprostenol has been limited due to its short half-life (3-5 minutes) and instability at room temperature. The use of Treprostinil, a more stable alternative drug, has provided PAH patients with more treatment options.
In 2021, clinical study data published in The Lancet Respiratory Medicine journal indicated that, for patients with interstitial lung disease and accompanying pulmonary arterial hypertension, inhaled Treprostinil improved patients' forced vital capacity (FVC) after 16 weeks compared to a placebo. This difference was most pronounced in patients with specific types of interstitial pneumonia, particularly those with Idiopathic Pulmonary Fibrosis. Inhaled Treprostinil appears to be a promising therapy for the treatment of Idiopathic Pulmonary Fibrosis.

In 2021, multiple teams jointly initiated a phase III clinical trial of Treprostinil for the treatment of idiopathic pulmonary fibrosis.

Nalbuphine is a κ-opioid receptor agonist and μ-opioid receptor antagonist that was originally researched and developed by Endo International plc. Currently, this medication is undergoing a Phase II clinical trial for Idiopathic Pulmonary Fibrosis (IPF).
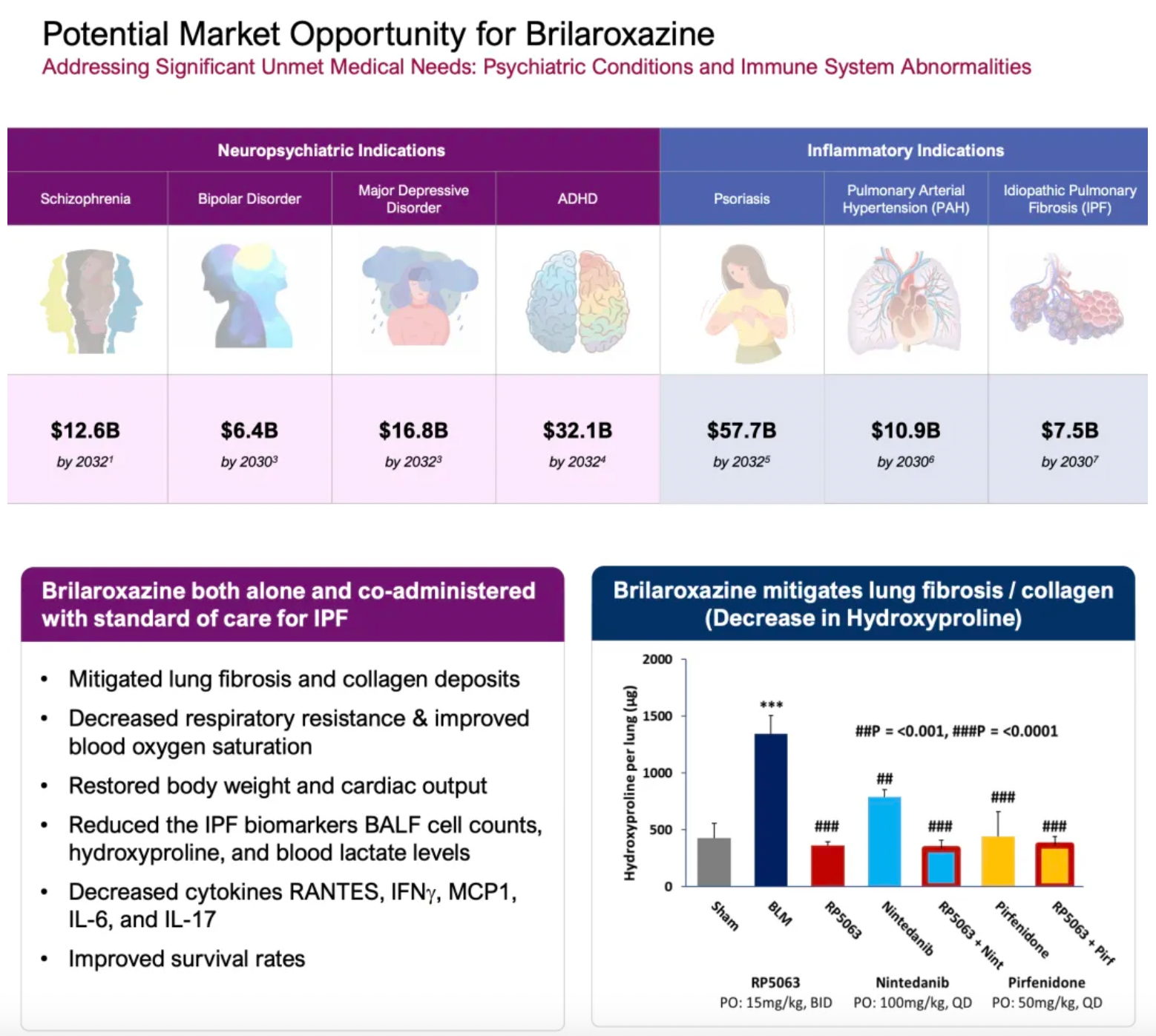
Brilaroxazine is a 5-HT1A/5-HT2A partial agonist originally researched and developed by Reviva Pharmaceuticals and is being used for the development of various clinical indications, including schizophrenia, idiopathic pulmonary fibrosis (IPF), and pulmonary arterial hypertension. Brilaroxazine has shown promising results in a Bleomycin-induced IPF rodent model when used alone or in combination with standard therapies. At present, this drug is also undergoing a Phase II clinical trial for IPF.
Setogepram (PBI-4050) is a novel orally bioavailable small molecule compound developed by Liminal BioSciences. Its mechanism of action includes being an agonist for GPR40 and an antagonist for GPR84, demonstrating anti-fibrotic activity in various fibrosis models, including pulmonary fibrosis. Data from a 12-week Phase 2 clinical trial published in 2019 evaluated the safety, efficacy, and pharmacokinetics of daily oral administration of 800 mg of PBI-4050 alone or in combination with nintedanib or pirfenidone in patients with mild to moderate idiopathic pulmonary fibrosis (IPF).

The 12-week open-label study investigated the effects of PBI-4050 in monotherapy in 9 patients, combined with nintedanib in 16 patients, and combined with nintedanib or pirfenidone in another 16 patients. The pharmacokinetic profiles were similar in both the PBI-4050 monotherapy group and the PBI-4050+nintedanib combination group, whereas the pharmacokinetics were reduced in the PBI-4050+pirfenidone group. There were no significant changes in Forced Vital Capacity (FVC) from baseline to week 12 in either the PBI-4050 monotherapy or the PBI-4050+nintedanib combination group. In contrast, a statistically significant decline in the percentage of predicted FVC was observed after 12 weeks of treatment with PBI-4050+pirfenidone. The stability of FVC between baseline and week 12 when using PBI-4050 alone or in combination with nintedanib was encouraging.




