Global First Drug Approvals in January 2025: A Comprehensive Review of Novel Therapies
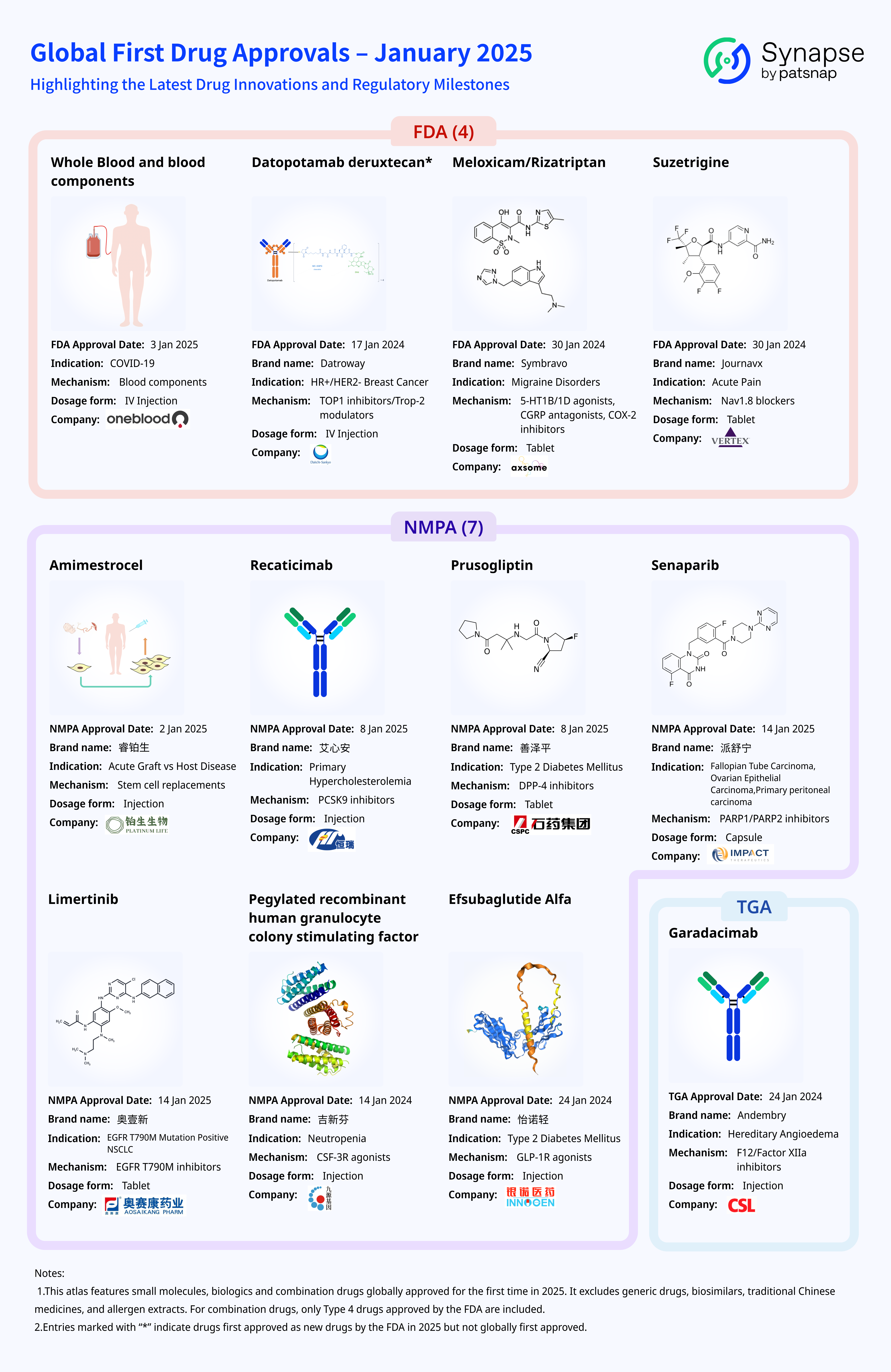
In this article, we present a comprehensive overview of drugs that received their first global approvals in January 2025, along with an analysis of these therapies. The drugs discussed in this article include small molecules, biologics, and combination drugs that were globally approved for the first time in January 2025. It is important to note that this compilation excludes generic drugs, biosimilars, traditional Chinese medicines, and allergen extracts. For combination drugs, only Type 4 drugs approved by the FDA are included.
Additionally, drugs that were first approved as new drugs by the FDA in January 2025 but had previously received approval in other countries are also included in this analysis and are marked with an asterisk (*) for clarity.
Which drugs were approved by the FDA?
1. Whole Blood and Blood Components
Whole Blood and Blood Components, specifically convalescent plasma derived from COVID-19 patients, received approval from the U.S. Food and Drug Administration (FDA) on January 3, 2025. The approval allows the use of high-titer SARS-CoV-2 antibody-containing whole blood-derived convalescent plasma and apheresis-derived convalescent plasma as a treatment for COVID-19 infection.
Developed and manufactured by OneBlood, Inc., COVID-19 convalescent plasma is human plasma collected from eligible blood donors by FDA-registered or licensed blood establishments. These donors must have recovered from symptomatic SARS-CoV-2 infection within the past six months and exhibit high levels of anti-SARS-CoV-2 antibodies in their plasma. The plasma’s neutralizing activity is determined based on donor antibody levels assessed through laboratory testing. Specifically, donor samples are evaluated using the GenScript cPass surrogate virus neutralization assay, which measures the inhibition of wild-type receptor-binding domain (RBD) binding to ACE2. The required inhibition rate must be equal to or greater than 80%.
This product is indicated for the treatment of COVID-19 in immunocompromised or immunosuppressed patients, particularly those at high risk of severe disease progression in both inpatient and outpatient settings. Convalescent plasma may be contraindicated in patients with a history of severe allergic reactions to plasma transfusion. The generally recommended dose is one unit (approximately 200 mL), with adjustments based on the patient's clinical response and physician’s discretion.
During administration, ABO blood type compatibility must be ensured, but Rh(D) compatibility is not required. Additionally, precautions should be taken to prevent container breakage or thawing issues during storage, and standard transfusion safety protocols should be followed to mitigate potential adverse effects and risks.
2. Datopotamab Deruxtecan*
Datopotamab deruxtecan is a Trop-2-targeting antibody-drug conjugate (ADC) jointly developed by Daiichi Sankyo and AstraZeneca. On December 27, 2024, Japan approved datopotamab deruxtecan for the treatment of adult patients with unresectable or metastatic hormone receptor-positive (HR+)/HER2-negative breast cancer who have received prior chemotherapy. This marked the drug’s first global marketing authorization. Subsequently, on January 17, 2025, the FDA also approved datopotamab deruxtecan for the treatment of unresectable or metastatic, previously treated HR+/HER2-negative breast cancer in adults. This approval was supported by data from the Phase III TROPION-Breast01 trial, which demonstrated a significant improvement in progression-free survival (PFS) and a reduction in the risk of disease progression or death compared to standard therapy.
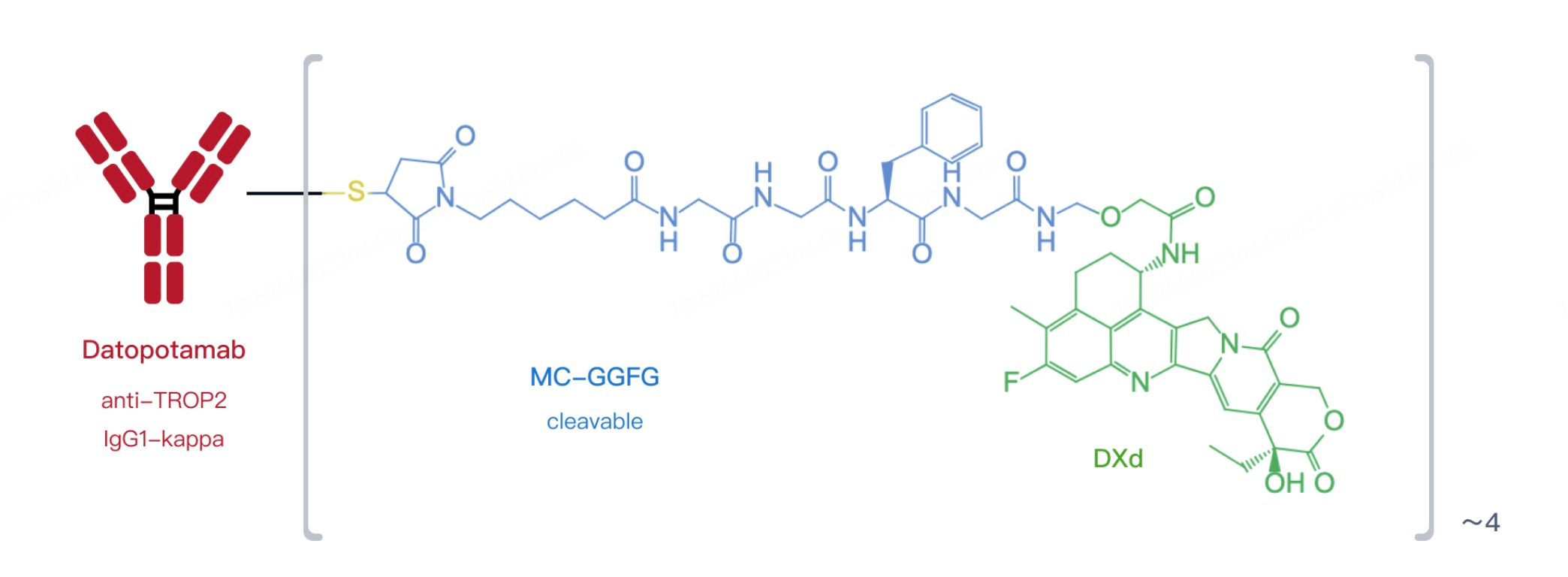
Datopotamab deruxtecan is designed to precisely target cancer cells expressing the Trop-2 antigen. Trop-2 is a protein widely expressed in various solid tumors, particularly breast and lung cancers. The drug consists of a humanized IgG1 monoclonal antibody conjugated to the potent topoisomerase I inhibitor deruxtecan (DXd). The antibody specifically binds to the Trop-2 antigen, while DXd functions as a cytotoxic chemotherapy agent that is released inside tumor cells, disrupting DNA replication and inducing cancer cell death. These components are linked by a cleavable tetrapeptide linker, ensuring stability in circulation and facilitating targeted drug release within tumor cells, thereby maximizing therapeutic efficacy.
Clinical trial results demonstrated that datopotamab deruxtecan provides significant efficacy in treating HR+/HER2-negative advanced breast cancer. In the pivotal Phase III TROPION-Breast01 study, the drug outperformed the standard therapy, docetaxel, in prolonging progression-free survival. With a median follow-up of 10.8 months, the median PFS in the datopotamab deruxtecan group was 6.9 months, compared to 4.9 months in the docetaxel group (HR = 0.63, 95% CI: 0.52–0.76, P < 0.0001), representing a 37% reduction in the risk of disease progression or death. Additionally, datopotamab deruxtecan demonstrated a high objective response rate (ORR), further reinforcing its potential as a novel cancer therapy. The drug also exhibited a favorable safety profile, with a lower incidence of grade ≥3 treatment-related adverse events compared to conventional chemotherapy, contributing to improved patient quality of life.
3. Meloxicam/Rizatriptan
The combination drug of meloxicam and rizatriptan, marketed under the brand name Symbravo, received approval from the FDA on January 30, 2025, for the treatment of migraines. This innovative small-molecule drug integrates two distinct mechanisms of action to address migraine symptoms effectively.
Developed by Axsome Therapeutics, Inc., meloxicam is a selective cyclooxygenase-2 (COX-2) inhibitor primarily used for pain and inflammation relief. Its high selectivity for COX-2 over COX-1 helps minimize the common gastrointestinal side effects associated with traditional nonsteroidal anti-inflammatory drugs (NSAIDs). By inhibiting the COX-2 enzyme, meloxicam reduces prostaglandin production, thereby alleviating pain.
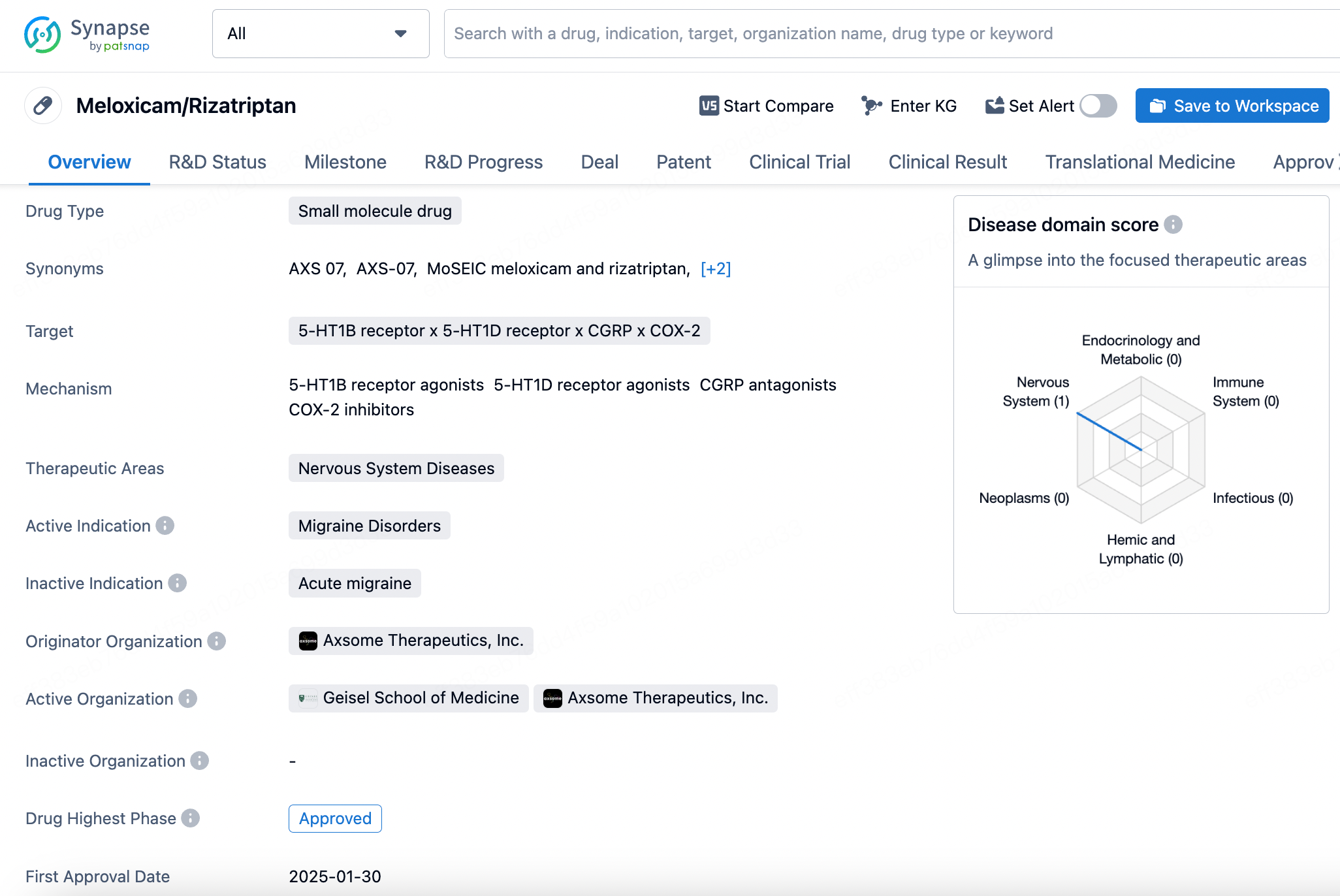
Rizatriptan, on the other hand, is a serotonin 5-HT1D and 5-HT1B receptor agonist, commonly used for the acute treatment of migraine attacks. It works by constricting dilated cerebral blood vessels and preventing the release of calcitonin gene-related peptide (CGRP) from nerve terminals—two key mechanisms believed to be crucial for migraine relief. Additionally, research suggests that rizatriptan may exert effects on the central nervous system, modulating specific receptors involved in pain signal transmission, further enhancing its analgesic efficacy.
The uniqueness of AXS-07 lies in its combination of these two active ingredients, aiming to leverage both the anti-inflammatory and analgesic properties of meloxicam and the migraine-specific physiological effects of rizatriptan. This dual-action approach provides a comprehensive treatment strategy, targeting both the pain and inflammation associated with migraines while addressing additional migraine-related symptoms. As a result, Symbravo offers patients more effective relief compared to traditional monotherapies.
4. Suzetrigine
Suzetrigine received approval from the FDA on January 30, 2025, as a small-molecule drug for the treatment of moderate to severe acute pain in adults. The drug's uniqueness lies in its selective targeting of the voltage-gated sodium ion channel Nav1.8, a channel specifically expressed in peripheral nociceptive neurons responsible for transmitting pain signals.
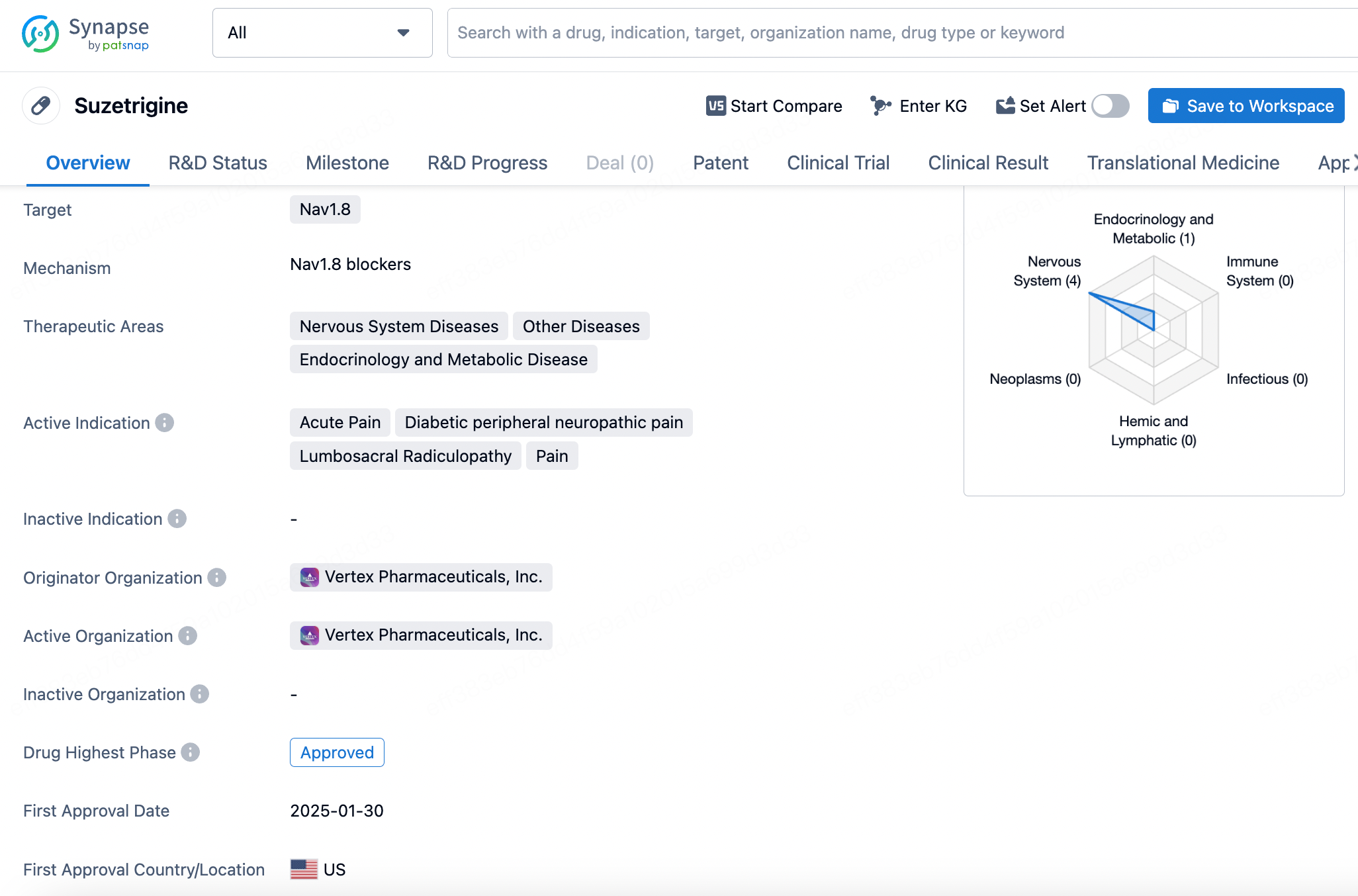
Developed by Vertex Pharmaceuticals, Inc., Suzetrigine is an X-type sodium channel α-subunit blocker that selectively inhibits Nav1.8 to prevent pain signal transmission. This high selectivity allows Suzetrigine to effectively relieve pain without affecting other types of sodium channels, thereby reducing the risk of side effects. Notably, Suzetrigine's mechanism avoids the addictive potential and adverse effects commonly associated with opioid analgesics, such as respiratory depression and constipation. This makes it a crucial new option for patients seeking non-opioid pain management solutions.
In the United States, over 80 million people require medication annually for managing moderate to severe acute pain, highlighting the urgent need for effective and non-addictive pain relief options. The successful development and approval of Suzetrigine mark the first market entry of a pain medication with a novel mechanism in nearly two decades. Clinical trials have demonstrated that Suzetrigine significantly reduced pain scores in patients undergoing abdominoplasty and bunionectomy, with no observed risk of addiction.
Suzetrigine represents a breakthrough in acute pain management, offering patients an innovative non-opioid, highly targeted analgesic option.
Which drugs were approved by the NMPA?
1. Amimestrocel
Amimestrocel is a human umbilical cord-derived mesenchymal stem cell (MSC) injection developed by Bosheng Excellence Biotechnology. On January 2, 2025, it received conditional approval from China’s National Medical Products Administration (NMPA) through the priority review and approval process, making it the first stem cell therapy to be approved for marketing in China.
Amimestrocel is indicated for the treatment of steroid-refractory acute graft-versus-host disease (aGVHD) with predominant gastrointestinal involvement in patients aged 14 and older. Graft-versus-host disease (GVHD) is a severe complication that may occur after allogeneic hematopoietic stem cell transplantation when donor immune cells recognize the recipient’s body as "foreign" and launch an immune attack.
A multicenter, randomized, double-blind, parallel, placebo-controlled Phase II clinical trial demonstrated that, in an intent-to-treat analysis, the MSC group exhibited superior therapeutic efficacy compared to the placebo group on Day 28. Additionally, clinical trial data showed that the therapeutic effects of Amimestrocel injection gradually became evident within an average of two weeks, with significant benefits observed particularly in patients with intestinal involvement.
Amimestrocel injection has shown promising efficacy in treating steroid-refractory aGVHD. According to clinical trial data, the treatment demonstrated a gradual therapeutic effect at a median of two weeks. Patients who completed eight infusions, especially those with gastrointestinal involvement, derived significant benefits from MSC therapy. These findings suggest that Amimestrocel may be particularly suitable for patients who have not responded well to conventional second-line treatments.
2. Recaticimab
Recaticimab received approval from the NMPA on January 8, 2025, marking its official entry into the Chinese market. Developed by Hengrui Medicine, Recaticimab is a humanized monoclonal antibody specifically targeting proprotein convertase subtilisin/kexin type 9 (PCSK9). PCSK9 primarily influences cholesterol metabolism by promoting the degradation of low-density lipoprotein receptors (LDLRs). Under normal conditions, LDLRs are located on the surface of hepatocytes, where they capture and remove low-density lipoprotein cholesterol (LDL-C) from the bloodstream. However, when PCSK9 binds to LDLRs, it triggers their internalization and subsequent degradation in lysosomes, thereby reducing the liver’s ability to clear LDL-C. Recaticimab specifically binds to PCSK9, preventing its interaction with LDLRs, thereby preserving LDLRs from degradation. This leads to an increased number of LDLRs on the hepatocyte surface, enhancing the body’s ability to clear LDL-C.
Recaticimab not only increases the number of LDLRs but also enhances their function. Since LDLRs play a key role in reducing serum LDL-C levels, increasing their quantity and activity is crucial for managing dyslipidemia. Additionally, Recaticimab utilizes "YTE" amino acid mutation technology to optimize its molecular structure, extending its half-life to approximately 27 days, significantly longer than other similar drugs. This allows for longer dosing intervals, such as every 4 or 8 weeks, improving patient adherence by reducing the inconvenience of frequent injections. This extended duration provides a more convenient option for the long-term management of hypercholesterolemia.
From a clinical perspective, Phase III clinical trial results demonstrate that Recaticimab significantly reduces LDL-C, total cholesterol (TC), non-high-density lipoprotein cholesterol (non-HDL-C), apolipoprotein B (ApoB), and lipoprotein(a) [Lp(a)], whether used as an adjunct to statins or as monotherapy. Moreover, it exhibits an excellent safety profile. Recaticimab is particularly suitable for patients who have poor responses to statins or are statin-intolerant, including those with primary hypercholesterolemia (both heterozygous familial and non-familial) and mixed dyslipidemia. The approval of Recaticimab brings new hope to the field of cardiovascular disease, offering significant benefits in improving patient quality of life and preventing cardiovascular events.
3. Prusogliptin
Prusogliptin received approval from the NMPA in China on January 8, 2025. Developed by CSPC Ouyi Pharmaceutical, Prusogliptin is a novel oral dipeptidyl peptidase-IV (DPP-4) inhibitor designed for the treatment of type 2 diabetes in adults. Its primary mechanism of action involves selectively and potently inhibiting DPP-4, thereby preventing the enzymatic degradation of glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). These two incretin hormones stimulate pancreatic β-cells to release insulin in response to food intake while simultaneously reducing α-cell secretion of glucagon, helping to regulate blood glucose levels. By inhibiting DPP-4, Prusogliptin prolongs the activity of GLP-1 and GIP, promoting postprandial insulin secretion, enhancing β-cell glucose sensitivity, and reducing unnecessary glucagon secretion, ultimately aiding in maintaining blood glucose levels within a healthier range.
Prusogliptin plays a crucial role in increasing the levels of endogenous GLP-1 and GIP. Under normal physiological conditions, these incretin hormones function as "intelligent" regulators of blood glucose, promoting insulin secretion only when glucose levels are elevated while avoiding excess insulin release under normal or low glucose conditions, thereby minimizing the risk of hypoglycemia. Moreover, unlike traditional insulin secretagogues that directly stimulate insulin secretion, Prusogliptin indirectly enhances endogenous GLP-1 function, making it less likely to cause hypoglycemia or lead to weight gain—important advantages for the long-term management of type 2 diabetes.
A study led by Peking University First Hospital demonstrated that patients in the Prusogliptin group experienced a significant reduction in glycated hemoglobin (HbA1c) levels compared to the placebo group, confirming its strong glucose-lowering efficacy. Additionally, Prusogliptin is suitable for monotherapy or combination therapy with metformin, making it particularly beneficial for patients who respond poorly to or cannot tolerate metformin. It is indicated for improving glycemic control in adult patients with type 2 diabetes, either as monotherapy or in combination with metformin. Furthermore, Prusogliptin exhibits sustained glucose-lowering effects, a low risk of hypoglycemia, and does not contribute to weight gain. It also has a favorable safety profile, with a low incidence of adverse reactions and minimal drug interactions. Notably, for patients with mild to moderate renal impairment, Prusogliptin does not require dose adjustment, further expanding its eligible patient population.
4. Senaparib
Senaparib was approved for market authorization in China on January 14, 2025. As a small-molecule chemical drug, Senaparib primarily targets poly (ADP-ribose) polymerase 1 and 2 (PARP1 and PARP2), exerting its anticancer effects by inhibiting the activity of these enzymes.
Developed by IMPACT Therapeutics, Senaparib operates based on the principle of synthetic lethality, playing a crucial role in the repair of single-strand DNA breaks. When these enzymes are inhibited, cancer cells with BRCA mutations or other homologous recombination repair deficiencies (HRD) are less able to effectively repair DNA damage, making them more prone to cell death. As a result, Senaparib is particularly suited for tumors that rely on PARP-mediated DNA repair pathways for survival.
According to the NMPA approval, Senaparib is indicated in China for the maintenance treatment of adult patients with advanced epithelial ovarian cancer, fallopian tube cancer, or primary peritoneal cancer following first-line platinum-based chemotherapy. This means that for patients who have achieved complete or partial remission after receiving first-line platinum-based chemotherapy, Senaparib provides a new maintenance treatment option aimed at prolonging progression-free survival and potentially improving overall survival outcomes.
The approval of Senaparib is based on the results of the FLAMES study, a randomized, double-blind, placebo-controlled, multicenter Phase III clinical trial. The results demonstrated that, compared to placebo, Senaparib significantly prolonged median progression-free survival (mPFS). Additionally, regardless of BRCA mutation status, patients derived benefits from the treatment. The drug also exhibited good tolerability and a manageable safety profile.
5. Limertinib
Limertinib is a novel small-molecule chemical drug that received marketing approval from the NMPA on January 14, 2025. It is a specifically designed inhibitor targeting the epidermal growth factor receptor (EGFR) T790M mutation.
As a third-generation EGFR tyrosine kinase inhibitor (EGFR-TKI), Limertinib selectively targets and inhibits EGFR proteins carrying the T790M mutation. This mutation commonly occurs in non-small cell lung cancer (NSCLC) patients who develop resistance after treatment with first- or second-generation EGFR-TKIs. By selectively binding to the EGFR T790M mutation site, Limertinib effectively blocks signal transduction pathways, thereby inhibiting tumor cell proliferation and spread.
In a pivotal Phase IIB clinical study, Limertinib demonstrated significant antitumor activity. A total of 301 patients with locally advanced or metastatic NSCLC who had progressed after prior EGFR-TKI therapy and tested positive for the EGFR T790M mutation participated in the study. The results, assessed by an independent review committee (IRC), showed an objective response rate (ORR) of 68.8%, a disease control rate (DCR) of 92.4%, a median duration of response (DoR) of 11.1 months, and a median progression-free survival (PFS) of 11.0 months. Furthermore, for patients with evaluable intracranial lesions, Limertinib also demonstrated promising efficacy, with an IRC-assessed best ORR of 65.9% and a median PFS of 10.6 months, indicating its potential effectiveness against central nervous system (CNS) metastases.
Limertinib is indicated for the treatment of adult patients with locally advanced or metastatic NSCLC who have progressed after prior EGFR-TKI therapy and have been confirmed to harbor the EGFR T790M mutation. This indication addresses a critical unmet need in treating resistance caused by specific genetic mutations, offering new hope to affected patient populations.
6. Pegylated Recombinant Human Granulocyte Colony-Stimulating Factor
Pegylated recombinant human granulocyte colony-stimulating factor (PEG-rhG-CSF) was approved for marketing in China on January 14, 2025, for the prevention and treatment of chemotherapy-induced neutropenia.
Developed by Hangzhou Jiuyuan Gene Engineering, PEG-rhG-CSF is a long-acting biologic drug designed to address chemotherapy-induced neutropenia. This drug is a conjugate of recombinant human granulocyte colony-stimulating factor (rhG-CSF) and polyethylene glycol (PEG), which extends the drug’s half-life in vivo and reduces the frequency of administration.
As a CSF-3R (colony-stimulating factor 3 receptor) agonist, PEG-rhG-CSF activates CSF-3R to stimulate the proliferation and differentiation of hematopoietic stem cells and progenitor cells in the bone marrow, leading to an increase in circulating neutrophils and enhancing the body's ability to resist infections. Compared to conventional rhG-CSF, PEG-rhG-CSF offers significant advantages, including prolonged duration of action and reduced injection frequency, which are crucial for improving patient adherence and quality of life. Additionally, the development of this drug addresses the clinical need for efficient, safe, and easy-to-manage treatment options.
7. Efsubaglutide Alfa
Efsubaglutide Alfa is an innovative fusion protein drug that was first approved by the NMPA of China on January 24, 2025. Developed by Guangzhou Yinnuo Pharmaceutical, Efsubaglutide Alfa is a glucagon-like peptide-1 receptor agonist (GLP-1RA). It is engineered as a recombinant protein by conjugating the GLP-1 receptor agonist to the Fc fragment of human immunoglobulin G2 (IgG2). This allows it to enhance insulin secretion in a glucose-dependent manner while inhibiting glucagon release, effectively controlling blood glucose levels in adult patients with type 2 diabetes.
What sets Efsubaglutide Alfa apart is its ultra-long-acting profile. The drug exhibits higher affinity for the GLP-1 receptor and has a prolonged degradation time in the body, with a half-life of 204 hours. This extended half-life enables a dosing regimen of once weekly or even once every two weeks, significantly improving patient convenience and treatment adherence. Additionally, clinical studies have shown that Efsubaglutide Alfa not only significantly lowers blood glucose levels but also improves lipid profiles, promotes weight loss, and offers potential cardiovascular protective effects. This makes it a comprehensive metabolic management option for patients with type 2 diabetes.
Which drugs were approved by the TGA?
Garadacimab
Garadacimab was first approved in Australia on January 24, 2025, as a novel treatment for hereditary angioedema (HAE). Developed by CSL Behring LLC, Garadacimab is a fully human IgG4 monoclonal antibody specifically designed to target and inhibit activated Factor XII (FXIIa), a key protein in the pathological process of HAE. By targeting and inhibiting FXIIa, the drug prevents excessive activation of the kallikrein-kinin system, thereby reducing the production of bradykinin, which is responsible for HAE symptoms. This mechanism of action provides patients with a long-acting and highly specific prophylactic treatment, significantly reducing the frequency of disease attacks and improving patients' quality of life.
Clinical studies, including the VANGUARD trial, have demonstrated that long-term prophylactic treatment with Garadacimab effectively controls acute HAE attacks. Compared to the placebo group, patients receiving Garadacimab reported a higher quality-of-life rating, indicating not only its medical superiority but also its positive impact on patients' overall well-being. Additionally, the drug is administered via subcutaneous injection once every 30 days, greatly enhancing treatment adherence and convenience for patients.
The approval of Garadacimab marks a significant advancement in HAE treatment, particularly in the field of prophylactic therapy. Unlike traditional C1 esterase inhibitor replacement therapy or direct blockade of the bradykinin B2 receptor, Garadacimab achieves therapeutic effects by modulating the upstream kallikrein-kinin pathway. This breakthrough not only provides HAE patients with a new treatment option but also showcases the potential of biotechnology in developing targeted therapies for specific molecular pathways.
How to obtain the latest research advancements in the field of biopharmaceuticals?
In the Synapse database, you can keep abreast of the latest research and development advances in drugs, targets, indications, organizations, etc., anywhere and anytime, on a daily or weekly basis. Click on the image below to embark on a brand new journey of drug discovery!