GPCR Family Anticancer Targets and Marketed Therapeutic Drugs (Part 2)



This article will continue to review the antitumor targets of the GPCR family and the therapeutic drugs that have been marketed.
03 CXCR4
The chemotactic properties of chemokines enable them to guide immune effector cells to sites of inflammation and coordinate interactions between immune cells. To date, approximately 50 human chemokines and 20 receptors have been identified.
Chemokines can be classified into two categories based on their function: inflammatory chemokines and homeostatic chemokines. They can also be categorized according to the position of the first two cysteine (C) residues in their protein sequence. There are four classes of chemokines: CC chemokines, CXC chemokines, C chemokines, and CX3C chemokines.
Taking chemokine CXCL12 (also known as stromal cell-derived factor-1, SDF-1) as an example, its interaction with the downstream receptor CXCR4 triggers downstream signaling pathways, impacting gene expression, cell chemotaxis, proliferation, and migration comprehensively. The CXCL12-CXCR4 signaling axis also plays a central role in tumor cell proliferation, angiogenesis, invasion, tumor microenvironment, and drug resistance induced by chemotherapy.
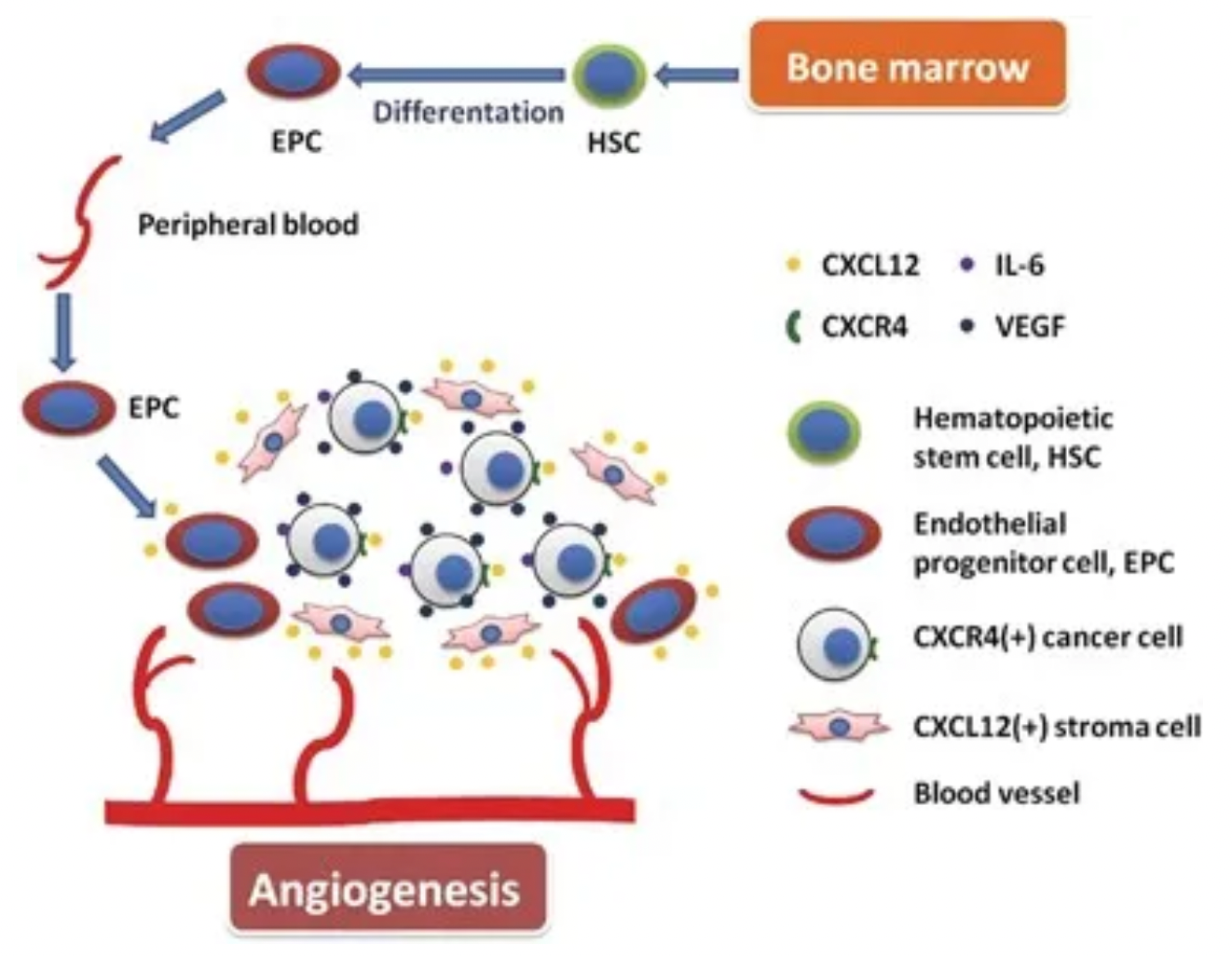
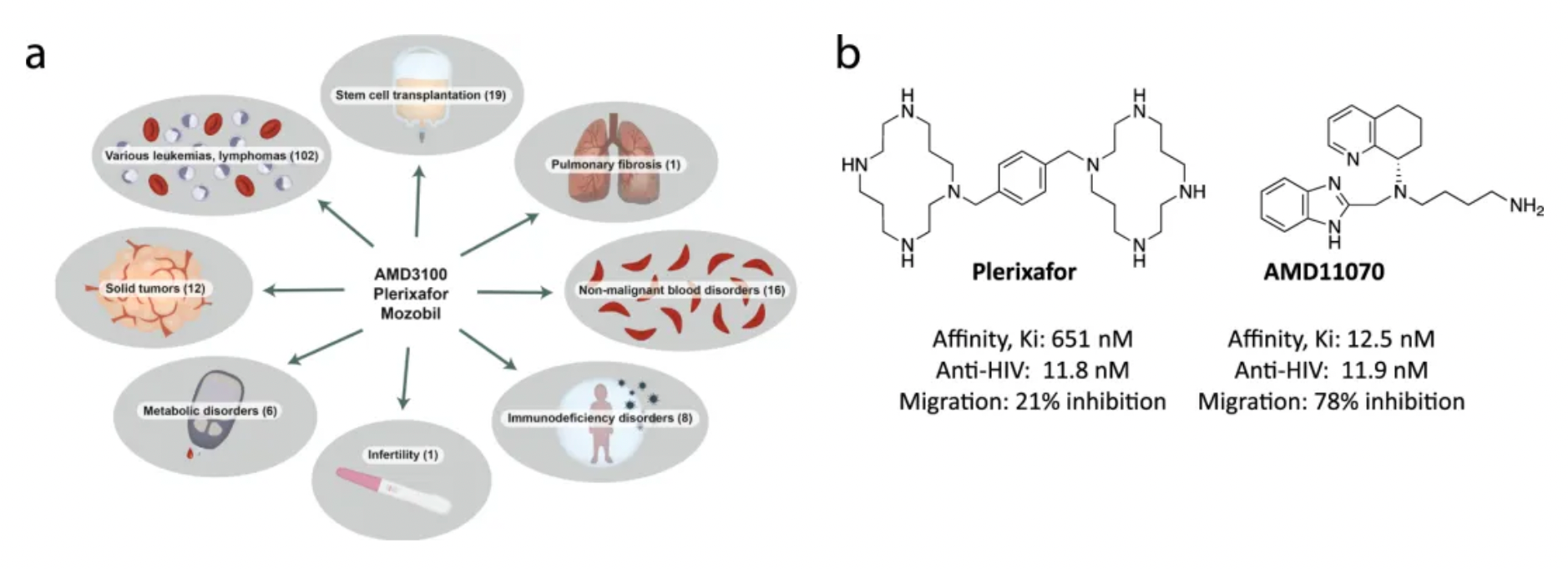
Plerixafor, developed by Genzyme Corp., is the first selective reversible CXCR4 antagonist small molecule compound. It is used in combination with granulocyte colony-stimulating factor (G-CSF) to mobilize hematopoietic stem cells from the bone marrow to peripheral blood, facilitating their collection for autologous transplantation in patients with non-Hodgkin's lymphoma and multiple myeloma. CXCR4 binding to its ligand SDF-1α is involved in the homing and transportation of CD34+ cells to the bone marrow. Plerixafor disrupts this interaction, leading to increased levels of CD34+ cells in peripheral blood. Plerixafor was granted orphan drug status in the USA and EU and received FDA approval in December 2008.
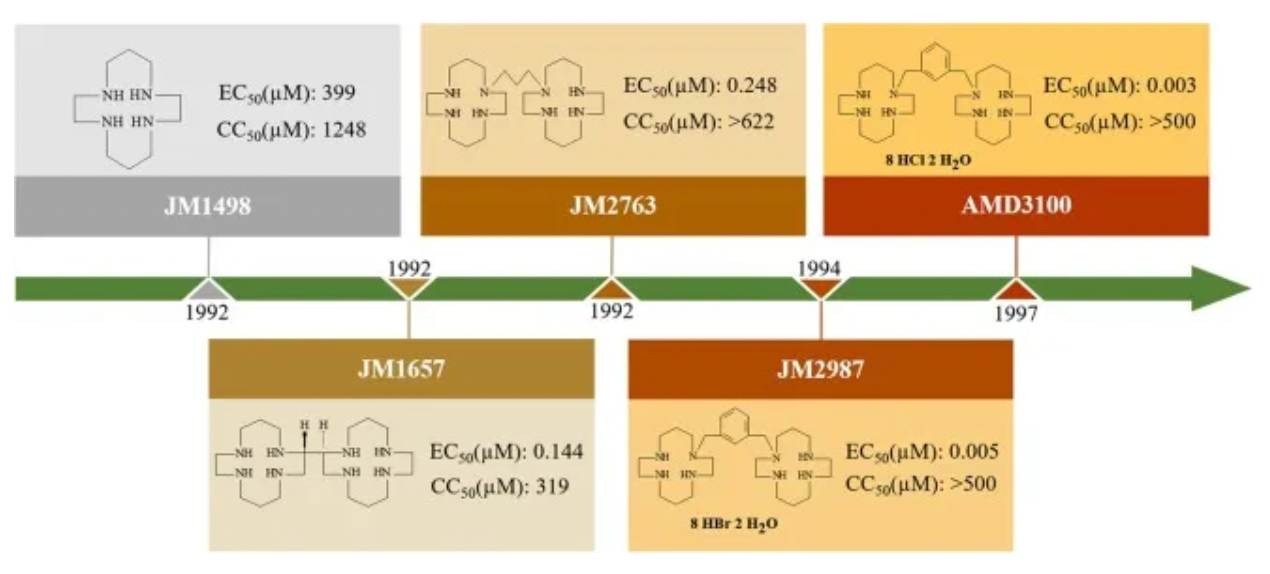
The development history of Plerixafor originated from initial attempts to develop therapeutic drugs for HIV. After a series of optimizations and modifications, Plerixafor demonstrated strong selectivity for CXCR4. However, Plerixafor's competitive binding ability to CXCL12 remains weak (Ki approximately 650nM).
Nevertheless, Plerixafor may induce severe hypersensitivity reactions such as anaphylactic responses. When used in leukemia patients, it can cause mobilization of tumor cells, splenic enlargement and rupture, embryotoxicity and fetotoxicity, and hematological effects such as leukocytosis and thrombocytopenia.
AMD11070 is a small molecule CXCR4 antagonist for oral use, originally developed by AnorMED. Initially, it was designed to combat HIV infections. AMD11070 specifically targets the X4 strain of HIV-1, which relies on the CXCR4 receptor. It has been shown that AMD11070 binds to the CXCR4 receptor and blocks its interaction with SDF-1α. Moreover, it inhibits calcium signaling induced by SDF-1, with an IC50 of 9.0 ± 2.0 nM, and inhibits SDF-1α-induced cell chemotaxis, with an IC50 of 19.0 ± 4.0 nM.
It is noteworthy that AMD11070 exhibits high selectivity for CXCR4, not inhibiting the downstream calcium signaling in cells expressing CXCR3, CCR1, CCR2b, CCR4, CCR5, or CCR7, nor does it inhibit ligand binding of CXCR7 or BLT1.
Subsequently, Genzyme Corporation undertook the development of AMD11070 (Mavorixafor) for various clinical indications over a span of 10 years. In April 2024, Mavorixafor was approved by the FDA in the USA for the treatment of WHIM syndrome and chronic neutropenia. Additionally, Mavorixafor is also being developed for clinical indications such as triple-negative breast cancer.
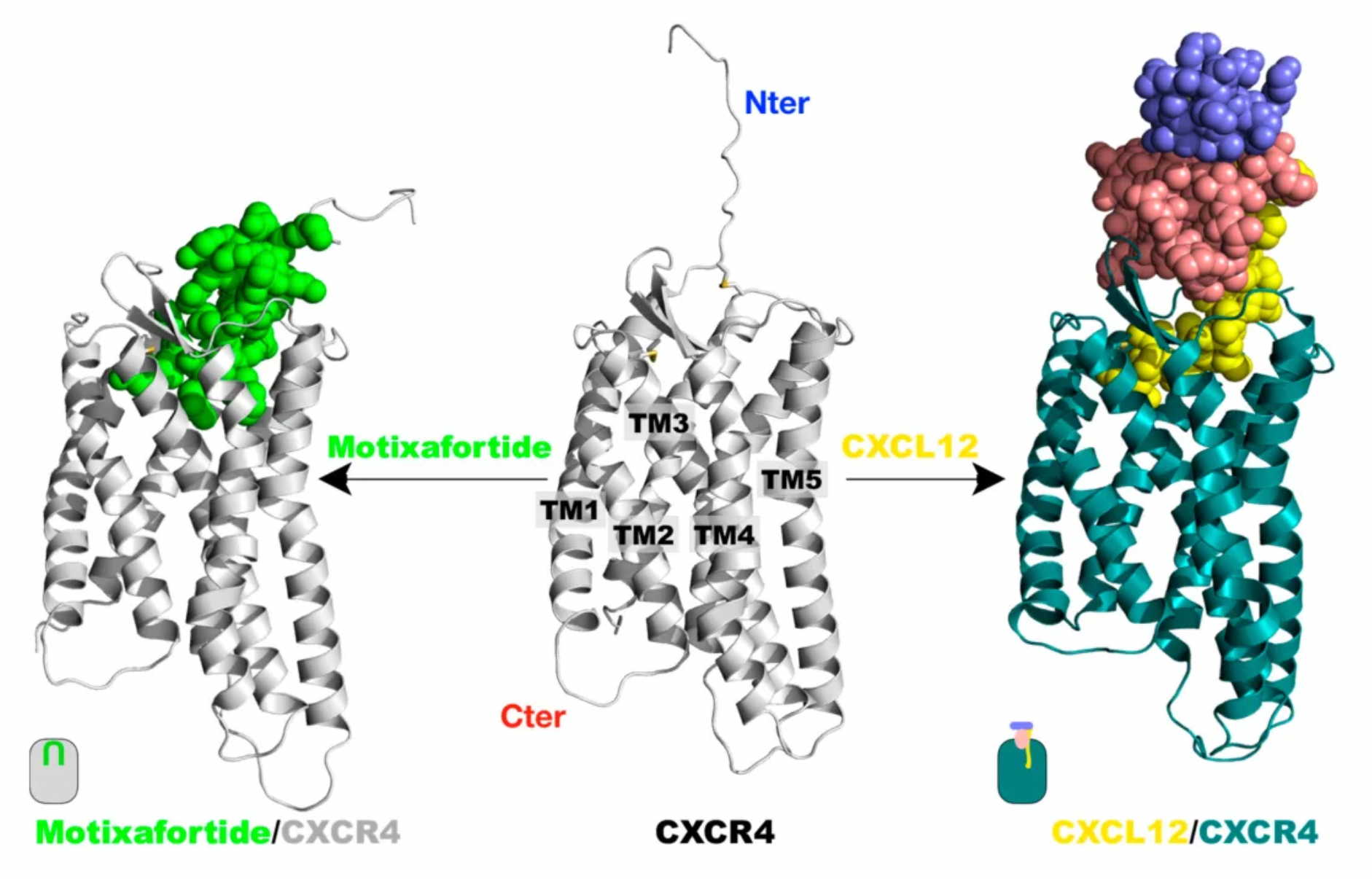
Motixafortide is another selective CXCR4 antagonist, developed by BioLineRx, for mobilizing hematopoietic stem cells (HSCs) and treating certain types of cancer. It was approved by the U.S. FDA on September 11, 2023, to be used in combination with Filgrastim (a granulocyte colony-stimulating factor, G-CSF) for mobilizing HSCs to peripheral blood, facilitating autologous transplantation for patients with multiple myeloma during a stem cell mobilization (SCM) process.
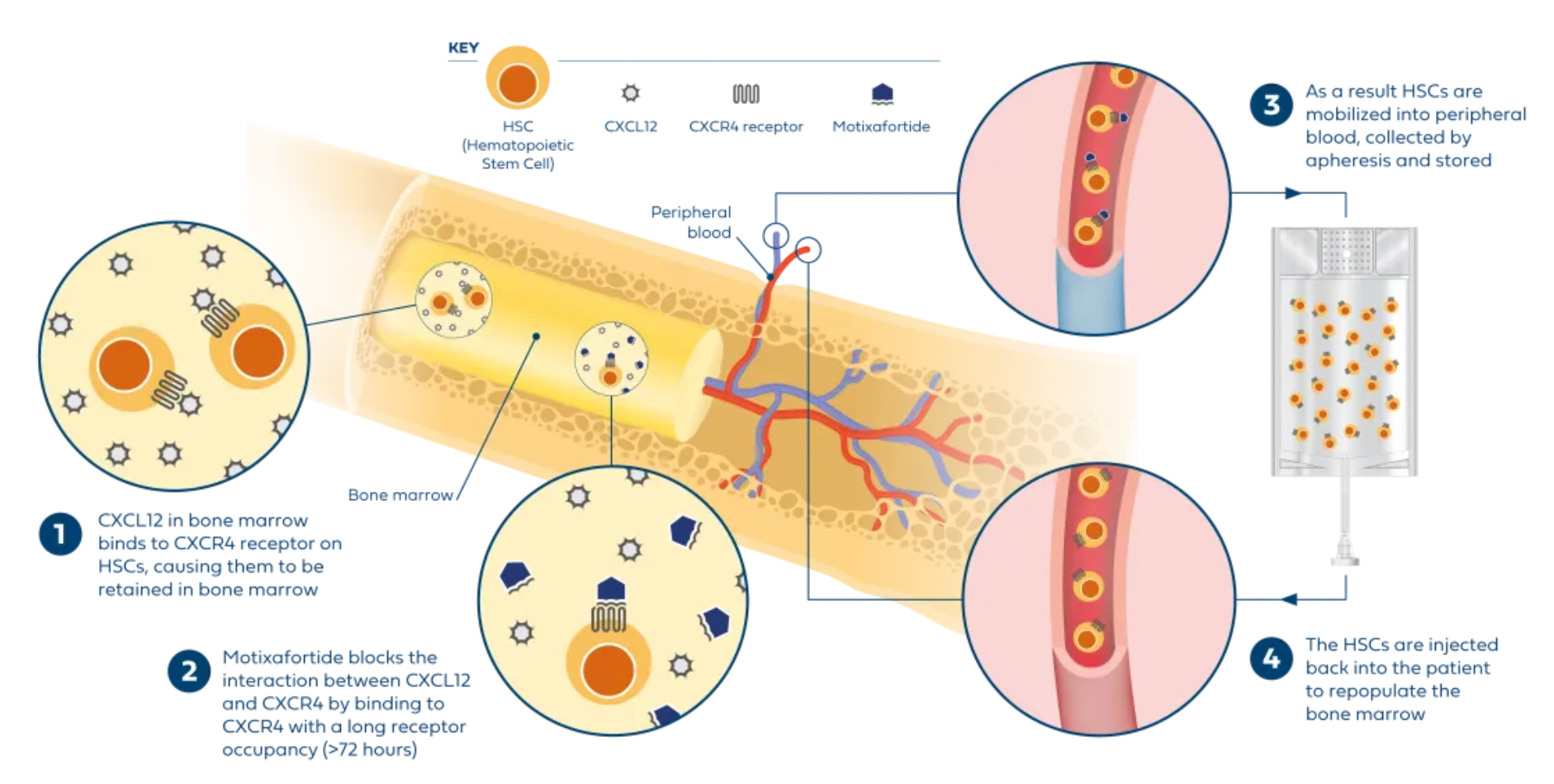
Regarding efficacy data: In two single-collection procedures, the combined treatment of Motixafortide with G-CSF enabled 67.5% of patients to achieve the target of ≥6 million CD34+ cells/kg per collection, compared to only 9.5% in the placebo plus G-CSF group. In terms of safety: The Motixafortide plus G-CSF regimen generally demonstrated good tolerability and safety profile. The most common adverse reactions (incidence >20%) in the GENESIS trial included injection site reactions (pain, erythema, and pruritus), pruritus, flushing, and back pain. Regarding other potential applications: Motixafortide has been granted orphan drug status in the EU and the USA for the treatment of pancreatic cancer and in the USA for the treatment of acute myeloid leukemia.
04 Histamine 2R (H2R)
Histamine is an endogenous biogenic amine that is widely distributed throughout the body, involved in many physiological and pathological conditions. Histamine exerts its effects through four histamine receptor subtypes (H1-H4) belonging to the GPCR family.
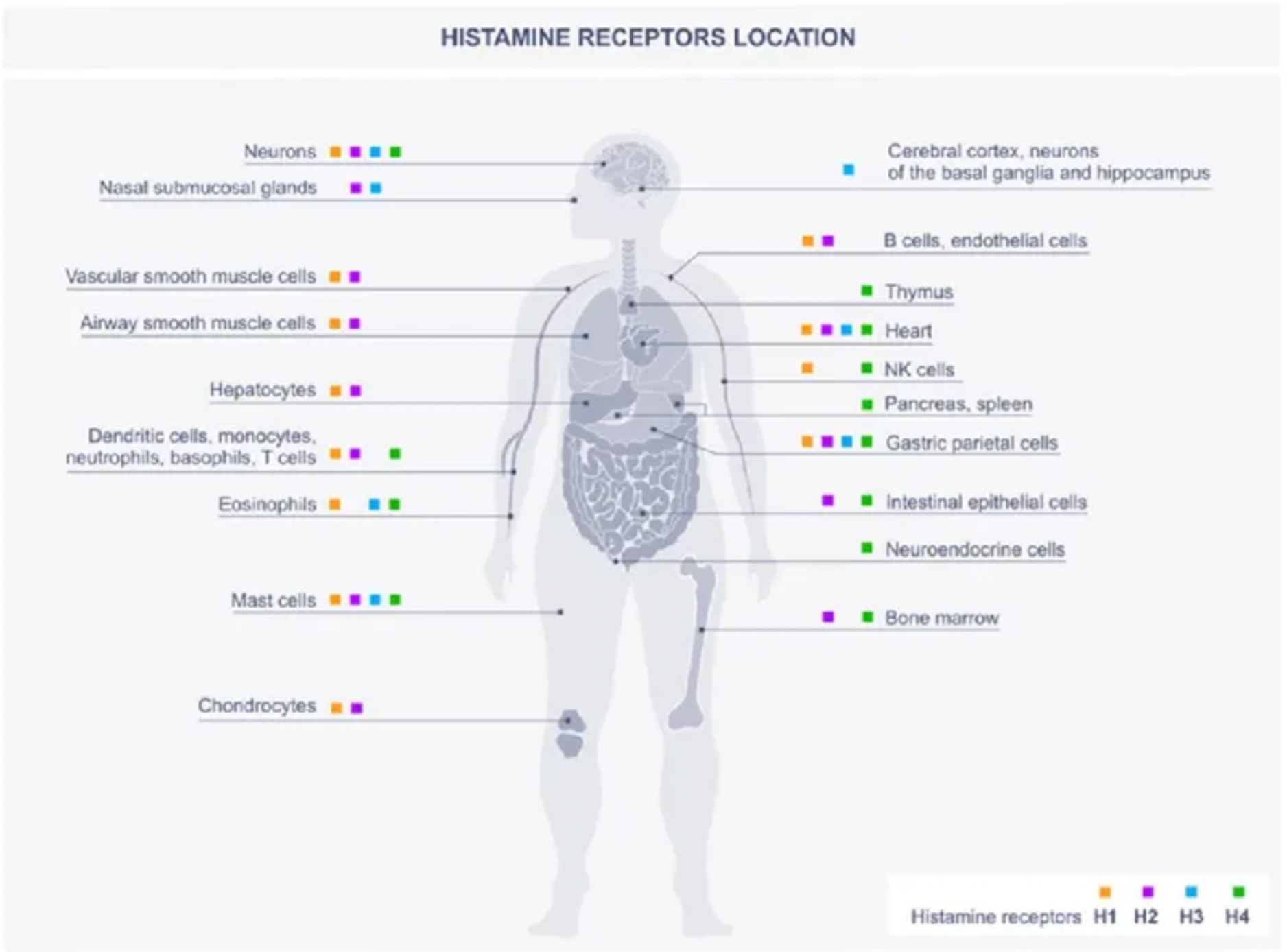
Histamine is one of the major mediators involved in inflammation and immune responses. The primary cellular sources of histamine include mast cells, basophils, histaminergic neurons, and enterochromaffin-like cells in the gastrointestinal tract, with smaller amounts also being released by epithelial cells or T cells. Additionally, high levels of histamine and its receptors in various types of cancers suggest their involvement in the complex biological processes of cancer. The expression of histamine receptors in multiple human cancer cell lines supports the role of histamine as an autocrine growth factor, which increases cell proliferation rates. Furthermore, histamine can stimulate various events associated with carcinogenesis, such as cell invasion, migration, and angiogenesis, demonstrating its critical role in cancer progression. Some HR antagonists, as well as inhibitors of histamine synthesis or transport, have been reported to have preventive effects on the growth of cancer cells.
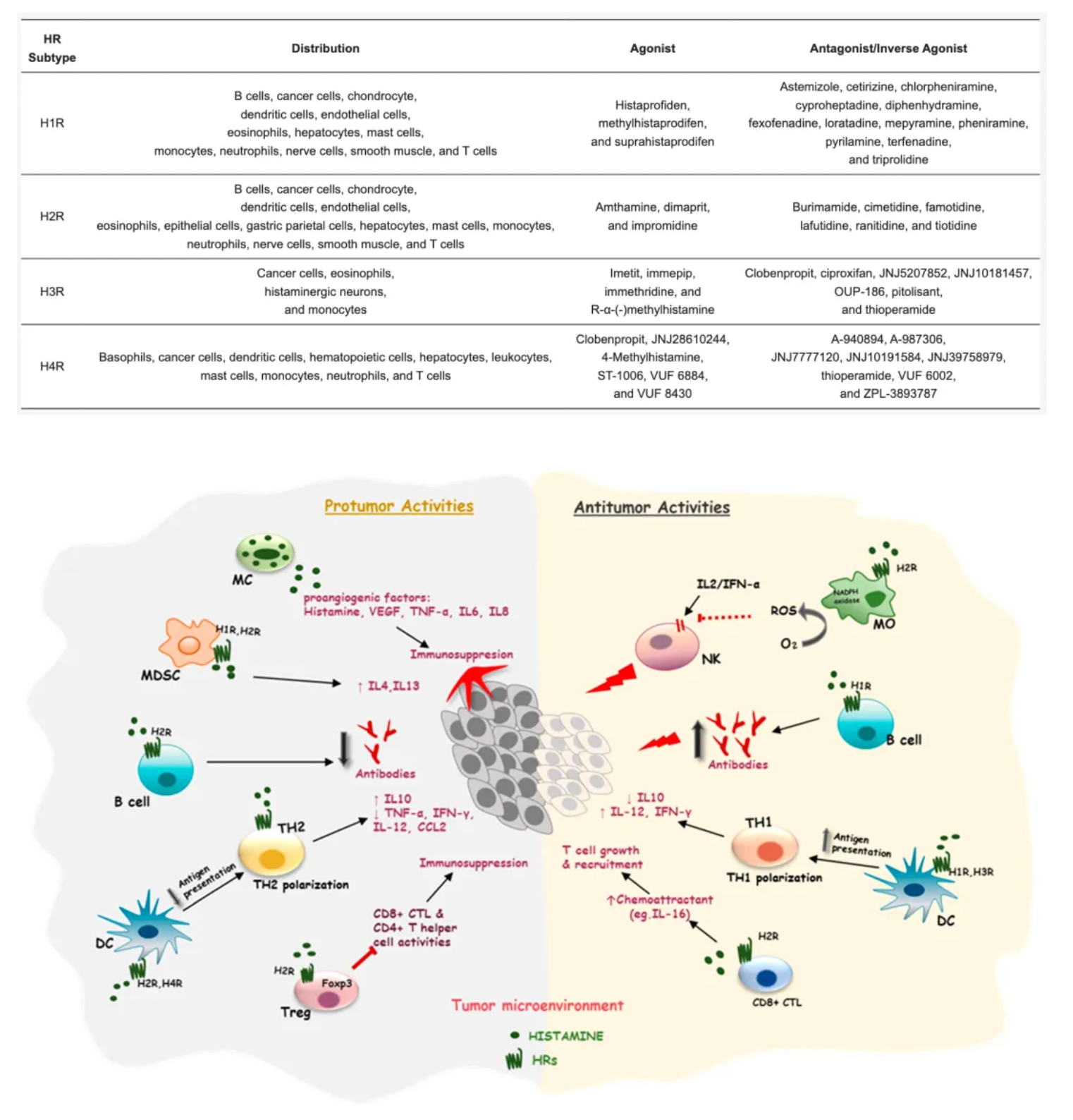
The H2 histamine receptor (H2R) plays an important physiological role in the human body, particularly in regulating gastric acid secretion. Activation of H2R can promote the production of stomach acid. Therefore, H2R antagonists, such as cimetidine, ranitidine, and famotidine, are widely used to treat diseases related to excess stomach acid, such as gastric ulcers and duodenal ulcers. Famotidine, developed later and approved by the FDA in 1985, has a higher affinity for the H2 receptor than cimetidine and ranitidine, potentially offering stronger gastric acid suppression. Famotidine is also used to treat gastric ulcers, duodenal ulcers, GERD, and can be used to prevent and treat upper gastrointestinal bleeding. Compared to cimetidine, famotidine has fewer drug interactions and milder side effects.
H2R antagonists are not first-line medications for cancer treatment. However, in cancer patients undergoing chemotherapy or radiotherapy, H2 receptor antagonists may be used to prevent or treat nausea and vomiting induced by the treatment, or to reduce the risk of upper gastrointestinal bleeding.
How to obtain the latest research advancements in the field of biopharmaceuticals?
In the Synapse database, you can keep abreast of the latest research and development advances in drugs, targets, indications, organizations, etc., anywhere and anytime, on a daily or weekly basis. Click on the image below to embark on a brand new journey of drug discovery!